Нейрофиброматоз I типа: причины, диагностика, лечение
Этиология и встречаемость нейрофиброматоза I типа. Нейрофиброматоз I типа (NF1, MIM №162200) — панэтническое аутосомно-доминантное заболевание с симптомами, чаще проявляющимися в коже, глазах, скелете и нервной системе. NF1 вызван мутациями в гене нейрофибромина (NF1). Болезнь встречается с частотой 1 на 3500 человек, что делает ее одним из наиболее частых аутосомно-доминантных заболеваний.
Приблизительно половина пациентов имеет новые мутации; частота мутаций в гене NF1 — одна из самых высоких среди всех генов человека, приблизительно 1 мутация на 10 000 живых новорожденных. Около 80% новых мутаций отцовского происхождения, однако влияние отцовского возраста на частоту мутации не подтверждено.
Патогенез нейрофиброматоза I типа
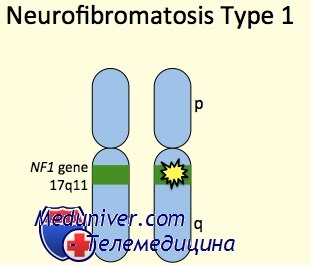
NF1 — большой ген (350 килобайт, 60 экзонов), кодирующий нейрофибромин, белок, широко экспрессируемый почти во всех тканях, но наиболее обильно в головном и спинном мозге и периферической нервной системе. Считают, что нейрофибромин регулирует несколько внутриклеточных процессов, в том числе активацию ГТФазы Ras, управляя клеточным делением и функционируя как супрессор опухолевого роста.
В гене NF1 описано более 500 мутаций, большинство из которых уникальны для каждой семьи. Клинические проявления связаны с потерей функции продукта гена; 80% мутаций вызывает укорочение молекулы белка. Мутация как причина болезни может быть выявлена более чем у 95% больных нейрофиброматозом I типа.
Нейрофиброматоз I типа характеризуется крайней клинической изменчивостью, как меж-, так внутрисемейной. Эта изменчивость, вероятно, вызвана комбинацией генетических, негенетических и случайных факторов. Отчетливой корреляции генотип-фенотип не обнаружено, хотя большие делеции чаще наблюдают у больных с неврологическими проблемами.
Фенотип и развитие нейрофиброматоза I типа
Нейрофиброматоз I типа — мультисистемное заболевание с неврологическими, мышечно-скелетными, глазными и кожными аномалиями, а также предрасположенностью к новообразованиям. Диагноз нейрофиброматоз I типа может быть поставлен, если обнаружены два или более следующих критериев: шесть или более пятен цвета «кофе с молоком», имеющие в диаметре, по крайней мере, 5 мм (до пубертата) или 15 мм (после пубертата); две или больше нейрофибромы любого типа или одну плексиформную нейрофиброму; веснушки в подмышечной или паховой области; глиому зрительного нерва; два или более узелков Лиша; характерный костный фенотип (сфеноидная дисплазия и уменьшение коры длинных трубчатых костей как с, так и без псевдоартрозов); наличие родственника первой степени родства с нейрофиброматозом I типа.
Почти все больные с нейрофиброматозом I типа без семейного анамнеза не имеют клинических критериев ранее 8 лет. Детям, унаследовавшим нейрофиброматоз I типа, диагноз обычно может быть установлен клинически уже на первом году жизни, так как для них требуется наличие только одного симптома болезни.
Хотя пенетрантность по существу полная, проявления чрезвычайно вариабельны. Множество пятен цвета «кофе с молоком» присутствуют почти у всех больных, веснушки наблюдают в 90% случаев. Множество больных с нейрофиброматозом I типа имеют только кожные симптомы болезни и узелки Лиша на радужке.
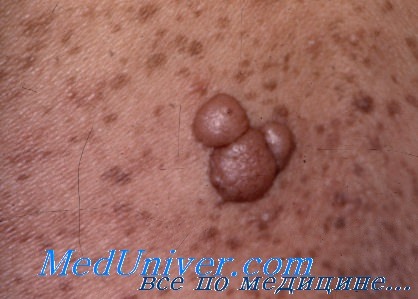
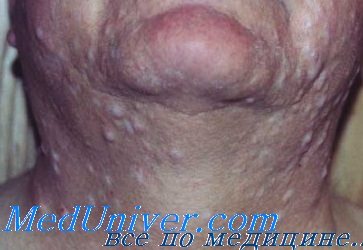
Многочисленные нейрофибромы обычно присутствуют у взрослых. Плексиформные нейрофибромы встречаются реже. Глазные проявления включают глиомы зрительного нерва (могут вести к слепоте) и узелки Лиша на радужке. Наиболее тяжелые осложнения со стороны костной системы — сколиоз, дисплазия позвонков, псевдоартрозы и избыточный рост. Часто встречаются также стеноз легочных, почечных и церебральных сосудов и гипертония. Наиболее частые опухоли у детей с нейрофиброматозом I типа (кроме нейрофибром) — глиомы зрительного нерва, опухоли мозга и злокачественные миелоидные болезни.
До половины всех детей с нейрофиброматозом I типа имеют когнитивные проблемы или дефицит внимания, сохраняющиеся во взрослом возрасте.
У больных с симптомами нейрофиброматоза I типа, ограниченными одной областью тела, имеющих здоровых родителей, может диагностироваться сегментный (или локальный) нейрофиброматоз I типа. Сегментный нейрофиброматоз I типа может представлять случайное необычное распределение клинических симптомов или соматический мозаицизм по мутации в гене NF1.
Особенности фенотипических проявлений нейрофиброматоза I типа:
• Возраст начала: от пренатального до позднего детства
• Пятна «кофе с молоком»
• Веснушки в подмышечных и паховых областях
• Нейрофибромы
• Узелки Лиша (гамартомы радужки)
• Плексиформные нейрофибромы
• Глиомы зрительного нерва
• Специфические повреждения костей
Лечение нейрофиброматоза I типа
Нейрофиброматоз I типа — клинический диагноз. Идентификацию мутаций в настоящее время стандартно не проводят из-за большого размера гена и крайней аллельной гетерогенности.
Эффективного лечения нет, следовательно, помощь сфокусирована на симптоматической терапии. Постоянное наблюдение пациентов с нейрофиброматозом I типа должно включать ежегодный медицинский осмотр, проводимый знакомым с этим заболеванием врачом, ежегодный осмотр окулиста в детстве (реже у взрослых), регулярную оценку психомоторного развития в детстве и регулярный контроль артериального давления.
Деформации, вызываемые нейрофиброматозом I типа, — наиболее беспокоящее проявление болезни. Отдельные кожные и подкожные нейрофибромы можно удалить хирургическим способом, если они доставляют косметические или другие неудобства. Плексиформные нейрофибромы, вызывающие обезображивание или беспокойства, также могут быть удалены хирургически. Тем не менее хирургическое вмешательство для этих неоплазм может быть проблематичным, так как они часто интимно спаяны с нервами и имеют тенденцию рецидивировать в месте резекции.
Риски наследования нейрофиброматоза I типа
Пациенты с нейрофиброматозом I типа имеют 50% риска родить ребенка, пораженного болезнью, хотя симптоматика у ребенка может отличаться от имеющейся в семье. Пренатальная диагностика доступна в семьях с известной мутацией в гене NF1, вызывающей болезнь, или информативных по сцеплению. Хотя пренатальный диагноз точен, он не обеспечивает прогностическую информацию из-за крайней фенотипической изменчивости болезни.
Родители больного ребенка, не имеющие никаких признаков болезни, имеют небольшое повышение риска повторения при следующей беременности из-за возможности полового мозаицизма, подтвержденного при нейрофиброматозе I типа.
Пример нейрофиброматоза I типа. Л.М., 2-летний мальчик, проходит обследование в связи обнаружением пяти пятен цвета «кофе с молоком», три из них более 5 мм в диаметре. У него не найдено веснушек в подмышечных или паховых областях, пороков развития кости и нейрофибром. Клинический осмотр обоих родителей не выявил признаков нейрофиброматоза. Генетик сообщил родителям и направившему педиатру, что не обнаружил клинических критериев для нейрофиброматоза I типа.
Ребенок повторно обратился в клинику генетики в 5 лет. У него появились узелки Лиша на радужке глаз и 12 пятен цвета «кофе с молоком», 8 из которых превышали 5 мм в диаметре. Обнаружена также веснушчатость в подмышечных областях с двух сторон. Установлен диагноз нейрофиброматоз I типа; родителям сообщили, что у ребенка новая мутация, следовательно, риск повторения низкий, но не исключен гонадный мозаицизм.
Родители отказались как от молекулярных анализов, так и от пренатальной диагностики при последующей беременности.