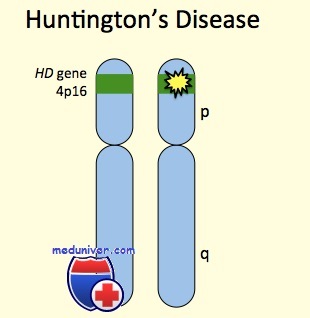
Этиология и встречаемость болезни Гентингтона. Болезнь Гентингтона (MIM № 143100) — панэтническое аутосомно-доминантное прогрессирующее нейродегенеративное заболевание, вызванное мутациями в гене HD. Распространенность болезни Гентингтона колеблется от 3 до 7 на 100 000 среди западных европейцев до 0,1—0,38 на 100 000 среди японцев. Эти изменения распространенности отражают изменения в распределении аллелей болезни Гентингтона и гаплотипов, предрасполагающих к мутации.
Патогенез болезни Гентингтона
Продукт гена HD, белок генгтинтин, экспрессируется повсеместно. Функция генгтинтина остается неизвестной.
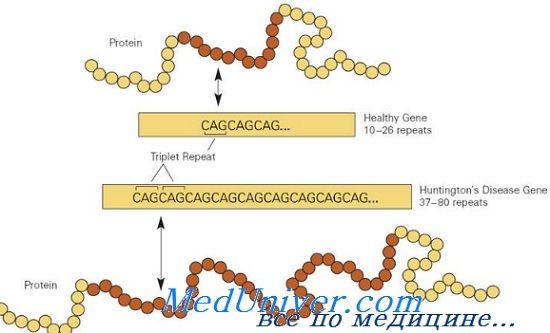
Болезнетворные мутации в гене HD обычно вызваны экспансией последовательности повторов CAG в экзоне 1, кодирующем полиглутаминовую цепочку; в норме аллели HD имеют от 10 до 26 повторов триплета CAG, тогда как мутантные аллели имеют больше 36 повторов. Приблизительно у 3% пациентов болезнь Гентингтона развивается в результате новой экспансии повторов CAG; 97% наследуют мутантный аллель HD от больного родителя.
Новые мутантные гены болезни Гентингтона возникают вследствие перехода премутации (27-35 повторов CAG) в полную мутацию. До настоящего времени все описанные пациенты наследовали новую полную мутацию от отца.
Экспансия полиглутаминового участка генгтинтина приводит к появлению у него нового свойства, и как необходима, так и достаточна для индукции болезни Гентингтона. Кроме выраженной рассеянной атрофии неостриатума, основного признака болезни Гентингтона, экспрессия мутантного генгтинтина вызывает дисфункцию нейронов, общую атрофию мозга, изменения уровней нейромедиаторов и накопление ядерных и цитоплазматических агрегатов в нейронах. В итоге экспрессия мутантного генгтинтина вызывает гибель нейронов; тем не менее похоже, что клинические симптомы и дисфункция нейронов предшествуют развитию внутриклеточных агрегатов и смерти нейронов. Механизм того, как экспрессия увеличенного числа глутаминовых остатков вызывает болезнь Гентингтона, остается неясным.
Фенотип и развитие болезни Гентингтона
Возраст начала болезни обратно пропорционален числу повторов CAG в гене HD. Пациенты с началом болезни во взрослом возрасте обычно имеют 40-55 повторов; в юношеском возрасте — обычно более 60 повторов. Пациенты с 36-41 повторами CAG имеют неполную пенетрантность, т.е. у них болезнь может не развиться в течение всей жизни. За исключением возраста начала, число повторов не влияет на другие проявления болезни Гентингтона.
Нестабильность и экспансия повторов CAG часто приводит к явлению антиципации, т.е. более раннему возрасту начала в последующих поколениях семьи. Достигнув однажды значения 36, число повторов увеличивается в ходе сперматогенеза отца; экспансия при материнской передаче мутации встречается реже. Поскольку число повторов триплета CAG обратно коррелировано с возрастом начала, индивидуумы, унаследовавшие мутацию от отца, имеют повышенный риск развития болезни с ранним началом; приблизительно 80% молодых пациентов наследуют мутантный ген болезни Гентингтона от отца.
Приблизительно треть пациентов имеют психиатрические нарушения; две трети — комбинацию познавательных и двигательных нарушений. Средний возраст пациентов на начало болезни — 35-44 года; приблизительно у четверти пациентов болезнь Гентингтона проявляется после 50 лет и у одной десятой — до 20 лет. Среднее выживание после установления диагноза — 15-18 лет, а средний возраст смерти — 54-55 лет.
Для болезни Гентингтона характерны прогрессирующие двигательные, познавательные и психиатрические расстройства. Двигательные нарушения включают как произвольные, так и непреднамеренные движения. Первоначально эти движения создают незначительные помехи ежедневной деятельности, но обычно приводят больного к нетрудоспособности по мере развития болезни.
Хорея, присутствующая у более чем 90% пациентов, — наиболее частая форма непроизвольного гиперкинеза; характеризуется неповторяющимися непериодическими толчками, которые не могут быть подавлены усилием воли. Познавательные нарушения начинаются в начале болезни и влияют на все аспекты познания; речь обычно затрагивается позднее, чем другие функции.
Поведенческие нарушения, обычно развивающиеся в ходе болезни, включают антиобщественное поведение, агрессию, взрывы гнева, апатию, сексуальные отклонения и повышенный аппетит. Психиатрические проявления, развивающиеся на любой стадии болезни, включают изменения личности, легкие психозы и шизофрению.
В конечных стадиях болезни Гентингтона обычно развиваются настолько выраженные двигательные нарушения, что больные полностью зависят от постороннего ухода. Они также теряют массу тела, у них появляются нарушения сна и мутизм. С течением болезни поведенческие нарушения уменьшаются.
Особенности фенотипических проявлений болезни Гентингтона:
• Возраст начала: от подросткового до старости
• Двигательные нарушения
• Познавательные нарушения
• Психиатрические нарушения
Лечение болезни Гентингтона
К настоящему времени никакого эффективного лечения при болезни Гентингтона нет. Помощь сводится к уходу и фармакологическому лечению поведенческих и неврологических проблем.
Риск наследования болезни Гентингтона
Каждый ребенок родителя с болезнью Гентингтона имеет 50% риск унаследовать мутантный аллель болезни Гентингтона. За исключением аллелей с неполной пенетрантностью (36-41 повтор CAG), у всех детей, унаследовавших мутантный аллель, если они имеют нормальную продолжительность жизни, развивается болезнь Гентингтона.
Дети отцов, несущих премутацию, имеют эмпирический приблизительно 3% риск унаследовать аллель болезни Гентингтона, в котором премутация перешла в полную мутацию. Тем не менее не все мужчины, несущие премутацию, равновероятно передают полную мутацию.
Доклиническое тестирование и пренатальная диагностика доступны благодаря анализу числа повторов CAG в 1 экзоне гена HD. Доклиническое тестирование и пренатальная диагностика — методики прогностические, поэтому лучше интерпретируются в случае подтверждения экспансии CAG у больного члена семьи.
Пример болезни Гентингтона. М.П., 44-летний мужчина, отметил ухудшение памяти и внимания. По мере снижения интеллектуальных функций в течение последующего года у него развились непроизвольные движения пальцев кистей и стоп, а также мимических мышц лица типа гримасничания. Он понимал свое состояние и впал в депрессию. До этого М.П. был здоров, и в его анамнезе не было аналогично больных родственников; его родители погибли в сорокалетнем возрасте в автокатастрофе. У пациента одна здоровая дочь.
После углубленного осмотра невролог диагностировал состояние больного как болезнь Гентингтона. Диагноз болезни Гентингтона подтвержден анализом ДНК, показавшим наличие 43 повторов триплета CAG в одном из аллелей гена HD (в норме не более 26). Последующее пресимптоматическое обследование дочери показало, что она также унаследовала мутантный аллель HD. Оба получили подробную консультацию.