Синдром Марфана имеет встречаемость около 1 на 10 000. Приблизительно 25-35% пациентов имеют новую мутацию. Мутации, вызывающие синдром Марфана, разбросаны по гену, и каждая мутация обычно уникальна в семье.
Патогенез синдрома Марфана
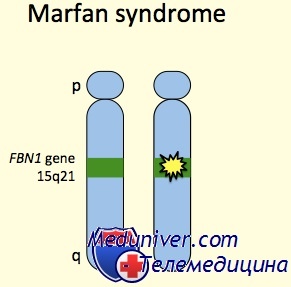
Ген FBN1 кодирует фибриллин 1, внеклеточный матричный гликопротеид с широким распределением. Фибриллин 1 полимеризуется, формируя микрофибриллы как в эластичных, так и в неэластичных тканях, например стенке аорты, цилиарных поясках и коже.
Мутации влияют на синтез, процессинг, секрецию, полимеризацию или устойчивость фибриллина 1. Исследования накопления и экспрессии фибриллина 1 в культуре клеток предположили доминантный отрицательный патогенез, т.е. синтез мутантного фибриллина 1 тормозит образование нормальных микрофибрилл нормальным фибриллином 1 или стимулирует протеолиз несоответствующих внеклеточных микрофибрилл.
Последние исследования на мышиных моделях синдрома Марфана указывают, что половинного количества нормального фибриллина 1 недостаточно, чтобы проводить эффективную сборку микрофибрилл. Таким образом, патогенезу болезни также может содействовать гаплонедостаточность.
Кроме синдрома Марфана, мутации в гене FBN1 могут вызывать другие синдромы, включая неонатальный синдром Марфана, изолированные скелетные симптомы, аутосомно-доминантную эктопию хрусталиков и фенотип MASS (марфаноидные симптомы, включая пролапс митрального клапана или миопию, пограничное и непрогресирующее расширение аорты, и неспецифические изменения скелета и кожи).
В общем, фенотипы довольно схожи в пределах семьи, хотя тяжесть фенотипических проявлений может значительно изменяться. До настоящего времени точное соотношение между генотипом и фенотипом не определено. Внутрисемейная и межсемейная изменчивость позволяет предполагать, что в определении фенотипа важную роль играют окружающая среда и эпигенетические факторы.
Последние исследования на мышиных моделях показывают, что фибриллин 1 не просто структурный белок, и что синдром Марфана не просто результат структурной слабости тканей. Более того, микрофибриллы фибриллина 1 в норме связывают и уменьшают концентрацию и активность факторов роста суперсемейства TGFb.
Потеря фибриллина 1 увеличивает сигналы свободного TGFb значительно содействующие заболеванию, так как антагонисты TGFb устраняют легочные и клапанные изменения, наблюдаемые у мышей с недостаточностью фибриллина 1.
Фенотип и развитие синдрома Марфана
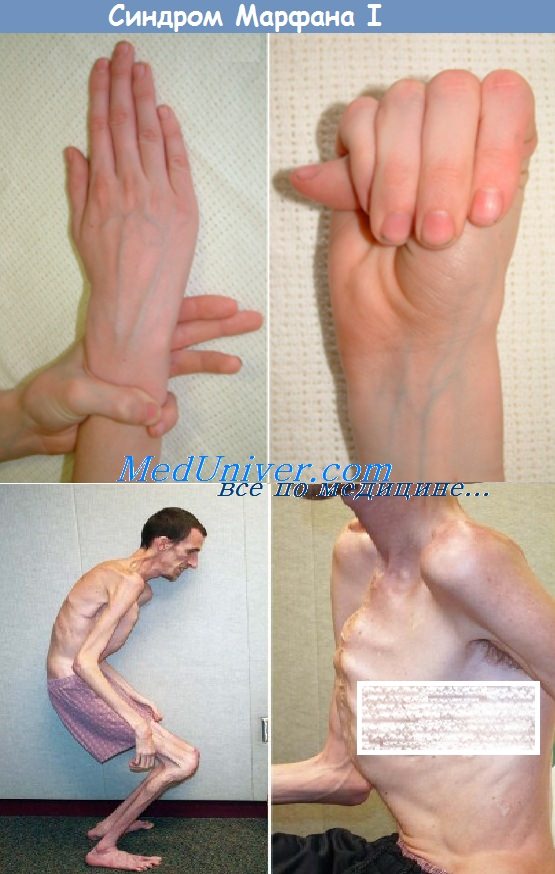
Синдром Марфана — мультисистемное заболевание со скелетными, глазными, сердечно-сосудистыми, легочными, кожными и другими аномалиями. Скелетные аномалии включают очень высокий рост (отношение размаха рук к росту >1,05; соотношение верхнего и нижнего сегментов <0,85 у взрослых), арахнодактилию, аномалии грудины, сколиоз, разболтанность суставов, готическое нёбо.
Аномалии глаз включают подвывих хрусталиков, уплощение роговицы, удлинение глазного яблока и гипоплазию радужки. Сердечнососудистые аномалии включают пролапс митрального клапана, аортальную регургитацию и расширение и расслаивающую аневризму восходящей аорты. Легочные аномалии включают спонтанный пневмоторакс и расширение концевых пузырьков. Аномалии кожи включают атрофические бороздки и рецидивирующие грыжи. Аномалии твердой мозговой оболочки включают выбухание оболочки в крестцово-поясничном отделе.
Большинство признаков синдрома Марфана появляются с возрастом. Скелетные аномалии типа аномалии грудины и сколиоза ухудшаются с ростом костей. Подвывих хрусталика часто присутствует уже в раннем детстве, но может развиваться и в юности.
С повышенной частотой при синдроме Марфана встречаются отслойка сетчатки, глаукома и катаракты. Сердечно-сосудистые осложнения обнаруживаются в любом возрасте и развиваются в течение всей жизни.
Основные причины преждевременной смерти пациентов с синдромом Марфана — сердечная недостаточность вследствие регургитации клапанов и аневризмы и разрыва аорты. Тем не менее в связи с улучшением хирургической и терапевтической помощи при аневризме аорты выживание улучшилось. С 1972 по 1993 г. ожидаемый возраст выживания для 50% пациентов поднялся с 49 до 74 лет для женщин и с 41 до 70 лет для мужчин.
Особенности фенотипических проявлений синдрома Марфана:
• Возраст начала: раннее детство
• Несоразмерно высокий рост
• Скелетные аномалии
• Эктопия (подвывих) хрусталика
• Пролапс митрального клапана
• Аневризма и разрыв аорты
• Спонтанный пневмоторакс
• Грыжа оболочки мозга в пояснично-крестцовом отделе
Лечение синдрома Марфана
Синдром Марфана — клинический диагноз, определяемый по наличию конкретных симптомов. Подтверждение синдрома Марфана идентификацией мутаций в гене FBN1 в настоящее время практически нецелесообразно, поскольку крайняя аллельная гетерогенность делает идентификацию причинно-обусловленной мутации в каждой семье крайне трудозатратной, а также из-за недостаточно надежной корреляции между генотипом и фенотипом. Анализ мутаций имеет недостаточную чувствительность или специфичность для синдрома Марфана, что ограничивает его клиническую пользу.
Для синдрома Марфана недоступно эффективное лечение; следовательно, помощь сфокусирована на профилактике осложнений и симптоматическом лечении. Оказание офтальмологической помощи включает регулярные осмотры, коррекцию миопии и, часто, замену хрусталика. Ортопедическая помощь заключается в укрепляющем лечении или хирургической коррекции сколиоза. Помощь при аномалии грудины в основном косметическая.
Физиотерапия может компенсировать нестабильность суставов. Сердечно-сосудистые проблемы решаются комбинацией терапевтических и хирургических мероприятий. Терапевтические усилия направлены на предохранение или замедление развития расширения корня аорты за счет уменьшения кардиологических показателей, снижения артериального давления и усилия выброса желудочков с помощью бета-адреноблокаторов, ограничение участия в контактных видах спорта, соревновательных видах спорта и в изометрических упражнениях.
Профилактическая замена корня аорты показана, когда расширение аорты или аортальная регургитация становится достаточно тяжелой. Большинству пациентов в настоящее время проводят надклапанную замену корня аорты, не требующую постоянного приема противосвертывающих препаратов.
Гемодинамические изменения, связанные с беременностью, могут приводить к прогрессирующему расширению или расслоению аорты. Полагают, что расслоение аорты вторично к гормональным изменениям, увеличению объема крови и сердечного выброса, связанных с беременностью и родами. Современные исследования считают, что риск беременности слишком велик, если ширина корня аорты превышает 4 см. Женщины могут выбрать проведение надклапанной замены аорты перед беременностью.
Риски наследования синдрома Марфана
Пациенты с синдромом Марфана имеют 50% риск иметь ребенка, больного синдромом Марфана. В семьях, передающих синдром Марфана, членов семьи, находящихся в группе риска, можно выявлять, либо обнаруживая мутацию (в тех редких случаях, когда она известна), либо анализом сцепления, если маркеры, тесно сцепленные с локусом FBN1, имеют очевидную связь с болезнью в семье пробанда. Пренатальная диагностика доступна только для семей, в которых возможны исследования сцепления или известна мутация в гене FBN1.
Пример синдрома Марфана. Д.Л., здоровый 16-летний ученик средней школы, звезда баскетбола, направлен в генетическую клинику для обследования по поводу синдрома Марфана. Телосложение Д.Л. похоже на телосложение его отца. Отец Д.Л., высокий субтильный человек, умер во время утренней пробежки; у других членов семьи случаев скелетных аномалий, внезапной смерти, снижения зрения или врожденных аномалий не было.
При медицинском осмотре выявлены астеническое телосложение, высокое дугообразное нёбо, небольшая деформация грудины по типу «куриной» груди, арахнодактилия, соотношение размах рук/рост 1,1, диастолический шум и стрии на плечах и бедрах. Эхокардиография выявила расширение корня аорты с аортальной регургитацией. Офтальмологическое обследование показало двусторонний иридодонез и легкое смещение хрусталиков кверху. На основе медицинского осмотра и результатов обследования генетик объяснил пациенту и его матери, что у него синдром Марфана.