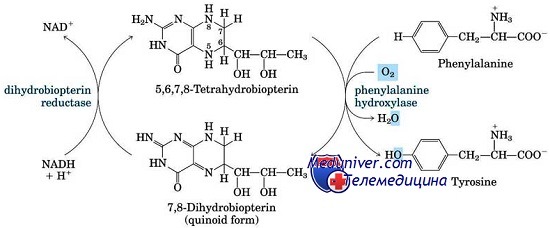
Аномалии, приводящие к увеличению уровня фенилаланина крови, чаще всего недостаточность фенилаланингидроксилаза (ФАГ) или фенилкетонурия (ФКУ), иллюстрируют почти все принципы биохимической генетики, относящиеся к дефектам ферментов. Все генетические аномалии метаболизма фенилаланина — следствие мутаций со снижением функции в гене, кодирующем ФАГ, или в генах, необходимых для синтеза или восстановления ее кофактора, ВН4.
Классическую фенилкетонурию (ФКУ) по праву считают образцовым представителем врожденных ошибок метаболизма. Это аутосомно-рецессивное заболевание распада фенилаланина, вызванное мутациями в гене, кодирующем ФАГ, фермент, преобразующий фенилаланин в тирозин. Открытие фенилкетонурии (ФКУ) Фелингом в 1934 г. впервые продемонстрировало генетический дефект как причину умственной отсталости.
Из-за неспособности к утилизации фенилаланина пациенты с фенилкетонурией (ФКУ) накапливают эту аминокислоту в жидкостях тела. Гиперфенилаланинемия повреждает формирующуюся в раннем детстве ЦНС и создает помехи функционированию зрелого мозга. Небольшая часть фенилаланина метаболизируется по альтернативным путям, производя повышенные количества фенилпировиноградной кислоты (кетокислота, по которой названа болезнь) и других метаболитов, выделяющихся с мочой.
Любопытно, что хотя ферментный дефект известен уже десятилетия, точный патогенетический механизм, каким образом увеличение фенилаланина повреждает мозг, все еще неизвестен. Важно, что развитие неврологического ущерба, вызванного метаболическим блоком при классической ФКУ, может в основном предупреждаться изменениями диеты, предохраняющими от накопления фенилаланина. Лечение фенилкетонурии (ФКУ) стало образцом для лечения многих метаболических болезней, исходы которых могут улучшаться за счет предотвращения накопления субстрата фермента и его производных.
Скрининг новорожденных на фенилкетонурию (ФКУ)
Широко используется популяционный скрининг новорожденных на фенилкетонурию (ФКУ). Фенилкетонурия (ФКУ) — образец генетических болезней, для которых оправдан массовый неонатальный скрининг; заболевание сравнительно часто встречается в ряде популяций (до 1 на 2900 живых новорожденных). Лечение, начатое в начале жизни, весьма эффективно; без лечения неизбежно развивается тяжелая умственная отсталость. Скрининг-тесты выполняют через несколько дней после рождения.
Капельку крови, полученную при проколе пятки, наносят на бумажный фильтр, высушивают и отправляют в централизованную лабораторию для оценки уровня фенилаланина в крови и соотношения фенилаланин/ тирозин. В прошлом образцы собирали перед выпиской ребенка из роддома. Тенденция к ранней выписке матери и новорожденного после родов изменила эту практику. Тест предпочтительно не делать до возраста 24 ч, поскольку уровень фенилаланина при фенилкетонурии (ФКУ) повышается только после рождения. Положительные результаты теста должны быть быстро подтверждены, поскольку задержка начала лечения более 4 нед после родов не позволяет избежать влияния на интеллектуальное состояние пациентов с фенилкетонурией (ФКУ).
Различные формы фенилкетонурии и гиперфенилаланинемия
Поскольку фенилкетонурия (ФКУ) связана с выраженной недостаточностью активности фенилаланингидроксилазы (ФАГ) (менее 1% по сравнению с контролем), мутантная ФАГ, имеющая остаточную активность, вызывает менее тяжелые фенотипические проявления, так называемую гиперфенилаланинемию и атипичную фенилкетонурию (ФКУ).
Гиперфенилаланинемию, отличную от фенилкетонурии (ФКУ), диагностируют, если концентрация фенилаланина в плазме ниже 1 ммоль/л на фоне нормальной диеты. Эта степень гиперфенилаланинемии только в 10 раз выше нормы и значительно ниже, чем концентрации, обнаруживаемые при классической фенилкетонурии (ФКУ) (>1 ммоль/л). Умеренное увеличение фенилаланина при гиперфенилаланинемии не способно повреждать функции мозга и может даже быть благоприятным, если увеличение небольшое (<0,4 ммоль), такие дети обращают на себя внимание врачей только благодаря скринингу. Их нормальный фенотип оказался наилучшим показателем безопасного уровня фенилаланина плазмы, который не следует превышать при лечении пациентов с классической фенилкетонурии (ФКУ).
Атипичная фенилкетонурия (ФКУ) — категория, включающая пациентов с промежуточным уровнем фенилаланина между классической ФКУ и гиперфенилаланинемией; такие пациенты требуют некоторого ограничения фенилаланина в диете, но меньшего, чем для пациентов с классической фенилкетонурии (ФКУ). Комплекс из этих трех клинических фенотипов с мутациями в гене ФАГ — пример клинической гетерогенности.
Гиперфенилаланинемии: аллельная и локусная гетерогенность при фенилкетонурии (ФКУ)
Молекулярные дефекты в гене фенилаланингидроксилазы. У пациентов с гиперфенилаланинемией, включая классическую фенилкетонурию (ФКУ), атипичную фенилкетонурию (ФКУ) и доброкачественные гиперфенилаланинемии, обнаружена поразительная степень аллельной гетерогенности в локусе фенилаланингидроксилазы (ФАГ) (более 400 различных мутаций по всему миру).
Подавляющее большинство аллелей фенилаланингидроксилазы (ФАГ) — достаточно редкие мутации, нарушающие ферментные свойства фенилаланингидроксилазы (ФАГ) и приводящие к гиперфенилаланинемии, хотя также обнаружены и доброкачественные полиморфизмы или менее частые доброкачественные варианты.
В популяциях европейского происхождения около двух третей известных мутантных хромосом представлены шестью мутациями. Шесть других мутаций ответственны за чуть более 80% мутаций фенилаланингидроксилазы (ФАГ) в азиатских популяциях. Остальные патогенные мутации встречаются реже. Чтобы сделать эту информацию широкодоступной, международным консорциумом разработана база данных мутаций в гене фенилаланингидроксилазы (ФАГ).
Во всех популяциях существует выраженная генетическая гетерогенность фенилаланингидроксилазы (ФАГ). Благодаря высокой степени аллельной гетерогенности в локусе, большинство пациентов с фенилкетонурией (ФКУ) во многих популяциях — компаундные гетерозиготы (т.е. у них присутствуют два разных патогенных аллеля), что полностью соответствует наблюдаемой ферментативной и фенотипической гетерогенности при нарушениях фенилаланингидроксилазы (ФАГ).
Сначала казалось, что знание генотипа фенилаланингидроксилазы (ФАГ) надежно предсказывает детали фенотипа; это ожидание оправдалось не полностью, хотя обнаружена определенная корреляция между генотипом ФАГ и биохимическим фенотипом.
В общих чертах мутации, которые полностью подавляют или резко снижают активность фенилаланингидроксилазы (ФАГ), вызывают классическую фенилкетонурию (ФКУ), тогда как мутации, приводящие к достаточно большой остаточной активности фермента, связаны с легкими фенотипами.
Тем не менее некоторые мутации фенилаланингидроксилазы (ФАГ) у гомозиготных пациентов определяют весь спектр фенотипов, от классической фенилкетонурии (ФКУ) до доброкачественной гиперфенилаланинемии.
Таким образом, стало очевидно, что в формировании фенотипа, наблюдаемого при специфическом генотипе, участвуют другие неопознанные биологические факторы, несомненно, включая гены-модификаторы. Это наблюдение, признанное в настоящее время общей характеристикой множества моногенных болезней, указывает на то, что даже моногенные болезни, подобные фенилкетонурии (ФКУ), — генетически не простые заболевания.
Дефекты в метаболизме тетрагидробиоптерина при фенилкетонурии (ФКУ)
Первоначально считали, что все дети с наследственной гиперфенилаланинемией имеют первичную недостаточность фенилаланингидроксилазы (ФАГ). Сейчас ясно, что примерно у 1-3% пациентов ген ФАГ нормален, а их гиперфенилаланинемия — результат генетического дефекта в одном из нескольких других генов, задействованных в синтезе или регенерации кофактора ФАГ, ВН4. Ассоциация одного фенотипа, например гиперфенилаланинемии, с мутациями в разных генах — пример локусной гетерогенности.
Как показывают мутации в генах, кодирующих белок фенилаланингидроксилазы (ФАГ) и метаболизм его кофактора биоптерина, белки, закодированные генами, демонстрирующими локусную гетерогенность, обычно входят в одну цепочку биохимических реакций. Пациенты с недостаточностью ВН4 сначала были выявлены из-за того, что, несмотря на успешное поддержание в диете низкой концентрации фенилаланина, у них рано развивались глубокие неврологические проблемы.
Плохие результаты частично объясняются необходимостью кофактора ВН4 для активности двух других ферментов, тирозингидроксилазы и триптофангидроксилазы. Обе этих гидроксилазы критичны для синтеза моноаминовых нейротрансмиттеров, таких как дегидроксифенилаланин, норэпинефрин, эпинефрин и серотонин. Пациенты с недостаточностью ВН4 имеют нарушение или в его биосинтезе из ГТФ, или в регенерации ВН4. Подобно классической фенилкетонурии (ФКУ), нарушение наследуется по аутосомно-рецессивному типу.
Очень важно отличать пациентов с дефектами в метаболизме ВН4 от больных с мутациями в фенилаланингидроксилазы (ФАГ), поскольку их лечение заметно разнится. Во-первых, так как белковая структура фенилаланингидроксилазы (ФАГ) у больных с нарушениями ВН4 нормальная, ее активность может восстанавливаться, если этим пациентам давать большие дозы ВН4, что приводит к снижению уровня фенилаланина плазмы. Следовательно, степень ограничения фенилаланина в диете пациентов с дефектами в метаболизме ВН4 может быть значительно уменьшена, а некоторые пациенты могут перейти на нормальную диету (т.е. без ограничения фенилаланина).
Во-вторых, необходимо также постараться нормализовать уровень нейротрансмиттеров в мозге этих пациентов, назначая продукты тирозингидроксилазы и триптофангидроксилазы: L-dopa и 5-гидрокситриптофан соответственно. По этим соображениям всем новорожденным с гиперфенилаланинемией показано обследование для определения аномалий в метаболизме ВН4.
Реакция на тетрагидробиоптерин при мутациях в гене ФАГ при фенилкетонурии (ФКУ)
У большинства пациентов с мутациями в гене фенилаланингидроксилазы (ФАГ), а не в метаболизме ВН4, отмечено отчетливое уменьшение уровня фенилаланина крови на фоне перорального приема больших доз кофактора фенилаланингидроксилазы (ФАГ) ВН4. Лучше отвечают на такое лечение пациенты со значимой остаточной активностью фенилаланингидроксилазы (ФАГ) (т.е. пациенты с атипичной фенилкетонурией (ФКУ) и гиперфенилаланинемией), но также поддается лечению небольшое число пациентов даже с классической фенилкетонурией (ФКУ). В то же время наличие остаточной активности ФАГ не дает гарантии влияния на уровень фенилаланина плазмы при назначении ВН4.
Наиболее вероятно, что степень ответной реакции на ВН4 зависит от специфических свойств каждого мутантного белка фенилаланингидроксилазы (ФАГ), отражающих лежащую в основе мутаций ФАГ аллельную гетерогенность. Показано, что введение в диету ВН4 оказывает лечебный эффект через несколько механизмов, вызванных повышением количества нормального кофактора, входящего в контакт с мутантным.
Эти механизмы включают стабилизацию мутантного фермента, защиту фермента от разложения клеткой, увеличение поступления кофактора к ферменту, имеющего низкое сродство с ВН4, и другие полезные эффекты в кинетических и каталитических свойствах фермента. Обеспечение повышенного количества кофактора — общая стратегия, применяемая в лечении многих врожденных ошибок метаболизма.
Материнская фенилкетонурия
Обычно успешное лечение фенилкетонурии (ФКУ) позволяет больным гомозиготам вести полноценную жизнь и иметь практически нормальные перспективы деторождения. В прошлом диету с низким содержанием фенилаланина прекращали у большинства пациентов с ФКУ в среднем детстве, основываясь на предположении (ошибочном, как установлено в настоящее время), что функционирование зрелой нервной системы не нарушается при возврате гиперфенилаланине-мии. Впоследствии было обнаружено, что почти все потомство женщин с фенилкетонурией (ФКУ), не получавших лечения, аномально; большинство этих детей с задержкой умственного развития, многие имеют микроцефалию, задержку роста и пороки развития, особенно сердца.
Как предсказывают принципы менделирующего наследования, все эти дети — гетерозиготы. Таким образом, задержка их развития вызвана не собственной генетической конституцией, а высокотератогенным эффектом высоких уровней фенилаланина в материнской крови. Соответственно необходимо, чтобы женщины с фенилкетонурией (ФКУ), планирующие беременность, начинали соблюдать диету с низким содержанием фенилаланина еще до зачатия.