Мукополисахариды или гликозаминогликаны — полисахаридные цепи, синтезируемые клетками соединительной ткани как нормальный компонент многих тканей. Они состоят из большого числа дисахаридных блоков; отличительная черта конкретного гликозаминогликана — природа двух молекул сахаров.
Разложение этих макромолекул происходит в лизосоме и требует пошагового удаления блока моносахаридов в конце цепи с помощью фермента, специфичного для моносахарида и вовлеченной цепи. Таким образом, для разложения любого гликозаминогликана необходима серия ферментов, и один фермент часто участвует в распаде более чем одного гликозаминогликана.
Мукополисахаридозы — разнородная группа, включающая более дюжины болезней накопления, при которых в результате недостатка одного из ферментов, необходимого для их разложения, в лизосомах накапливаются мукополисахариды. При конкретных мукополисахаридозах могут накапливаться один или несколько гликозаминогликанов, если отсутствует фермент, необходимый для их распада.
Недеградировавшие гликозаминогликаны появляются в моче, где они могут быть обнаружены скрининговыми тестами.
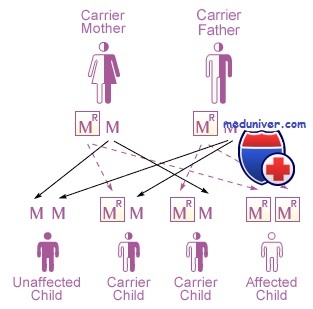
Первые два описанных мукополисахаридоза — Х-сцепленный рецессивный синдром Хантера и более тяжелый аутосомно-рецессивный синдром Гурлер. Каждое из этих заболеваний сначала называли гаргоилизмом из-за грубости черт лица больных детей. Больные дети имеют скелетные аномалии и низкий рост, умственную задержку, а также другие аномалии.
Синдром Гурлер — следствие выраженной недостаточности а1-идуронидазы. Клинически отличное заболевание — синдром Шейе — первоначально считали вызванным мутацией в другом локусе, в основном из-за значительно более мягкого фенотипа. Тем не менее синдромы Шейе и Гурлер оказались аллельными, а мутации а1-идуронидазы, вызывающие синдром Шейе, связаны с более высокой остаточной активностью.
Примеры мукополисахаридозов:
1. Гурлер. Диагностируется рано (<18 мес), помутнение роговицы, скелетные изменения, гепатоспленомегалия, грубое лицо, смерть до 10-летнего возраста. Дефект фермента: a-1-идуронидаза. Наследование: аутосомно-рецессивное. Из-за аллелей, которые существенно нарушают активность фермента.
2. Шейе. Начало после 5 лет жизн, нормальный интеллект и продолжительность жизни, роговичные помутнения, клапанные пороки сердца. Дефект фермента: a-1-идуронидаза. Наследование: аутосомно-рецессивный. Очевидно, из-за аллелей, которые оставляют некоторую остаточную активность фермента.
3. Хантера. Гурлер-подобный синдром, но с медленным течением. Дефект фермента: Идуронатсульфатаза. Наследование: Х-сцепленный рецессивный. Мягкий фенотип с переменным поражением цнс.
Разные типы наследования аутосомного синдрома Гурлер и Х-сцепленного синдрома Хантера указывают, что они возникли вследствие мутаций в разных генах. Это различие также показано на клеточных культурах. Хотя фибробласты пациентов при обеих болезнях накапливают мукополисахариды в культуральной среде, накопление корректируется при совместном культивировании обоих типов клеток в одной культуральной посуде.
Коррекция происходит вследствие захвата фибробластами с недостаточностью a-L-идуронидазы при синдроме Гурлер нормальной a-L-идуронидазы, синтезированной фибробластами синдрома Хантера; в клетках с синдромом Хантера происходил обратный феномен. Этот простой эксперимент оказался красивой иллюстрацией того, что две болезни повреждают разные белки. Явление, когда продукт генома одного мутантного гена может скорректировать биохимический дефект у другого, называется генетической комплементацией, а исследования, используемые для обнаружения генетической комплементации, называют анализом комплементации.
Способность клетки усваивать недостающий лизосомный фермент из внеклеточной жидкости — один из механизмов коррекции биохимического дефекта при болезнях накопления после пересадки пациентам нормальных клеток (выделяющих фермент). Радикальный терапевтический эффект получен при лечении некоторых пациентов с мукополисахаридозами, включая синдром Гурлер, при пересадке костного мозга.
Способность клеток усваивать лизосомные ферменты из внеклеточной жидкости также дает обоснование для заместительной ферментной терапии при многих болезнях — стратегия, в значительном числе случаях оказавшаяся в высшей степени эффективной.