Лизосомы — ограниченные мембранами органеллы, содержащие набор гидролитических ферментов, ответственных за деградацию многих биологических макромолекул. Генетические дефекты гидролаз приводят к накоплению их субстратов в лизосоме, вызывая клеточную дисфункцию и, в конечном счете, смерть клетки.
Постепенное накопление субстрата ответственно за одну общую клиническую характеристику таких болезней — их неуклонно прогрессирующее течение. При большинстве заболеваний накопление субстрата клинически обнаруживают как увеличение массы пораженных тканей и органов. Тем не менее, когда поражается мозг, как это нередко случается, результатом бывает одна из форм нейродегенерации.
Клинические фенотипы часто делают диагноз болезни накопления простым и обычно позволяют установить если не специфическое заболевание, то класс болезней накопления. Описано более 50 аутосомно-рецессивных заболеваний лизосомных гидролаз или нарушений транспорта мембраны лизосом. До недавних пор эти болезни не поддавались лечению. Однако появление метода заместительной ферментной терапии резко улучшило долгосрочный прогноз пациентов с некоторыми из этих заболеваний.
Болезнь Тея-Сакса — одна из группы разнородных лизосомных болезней накопления, GМ2-ганглиозидоз, вызванный невозможностью деградации сфинголипида, ганглиозида GM2. Биохимическое нарушение — выраженная недостаточность гексозаминидазы А. Хотя фермент встречается во всех тканях, болезнь имеет клиническое влияние почти исключительно на мозг, преобладающее место синтеза ганглиозида GM2.
Каталитически активная гексозаминидаза А — продукт системы из трех генов. Гены кодируют а- и b-субъединицы фермента (гены НЕХА и НЕХВ соответственно) и белок-активатор, соединяющийся с субстратом и ферментом, прежде чем фермент сможет расщепить терминальный остаток N-ацетил-b-галактозамина в ганглиозиде.
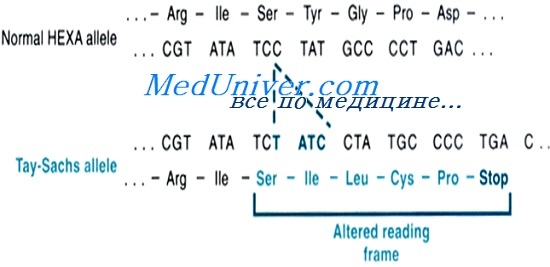
Клинические проявления дефектов в трех генах идентичны, но их можно различить при ферментном анализе. Мутации в гене НЕХА влияют на a-субъединицу и нарушают работу гексозаминидазы А, вызывая болезнь Тея-Сакса (или менее тяжелые варианты недостаточности гексозаминидазы). Аллели болезни Тея-Сакса ведут к выраженному недостатку мРНК a-субъединицы и недостаточности гексозаминидазы А. Дефекты в гене НЕХВ или гене, кодирующем белок-активатор, нарушают активность гексозаминидазы А и гексозаминидазы В, вызывая болезнь Сандхоффа и очень редкий дефицит белка-активатора соответственно.
Клиническое течение болезни Тея-Сакса особенно трагично. Пораженные дети рождаются здоровыми и нормально развиваются примерно до 3-6 мес жизни, затем постепенно развивается прогрессирующее неврологическое ухудшение, приводящее к смерти в возрасте от 2 до 4 лет. Эффекты гибели нейронов могут наблюдаться непосредственно в форме симптома так называемой «вишневой косточки» на сетчатке, представляющего собой выдающуюся центральную ямку, окруженную бледной макулой.
В отличие от этого, аллели НЕХА, приводящие к некоторой остаточной активности фермента, вызывают формы неврологической болезни с поздним началом или, в случае аллелей псевдонедостаточности (обсуждаемых позже), вообще не вызывают болезни. В варианты с поздним началом проявления обычно включают дисфункцию нейронов нижних конечностей и атаксию из-за спиноцеребеллярной дегенерации, но, в отличие от ранней детской формы, зрение и интеллект обычно остаются нормальными, хотя у трети пациентов развиваются психозы.
Популяционная генетика болезни Тея-Сакса
При многих моногенных болезнях некоторые аллели обнаруживаются с более высокой частотой в одних популяциях по сравнению с другими. Эту ситуацию иллюстрирует болезнь Тея-Сакса, при которой три аллеля составляют до 99% аллелей у евреев ашкенази, в то время как в других популяциях два других аллеля, ни один из которых не встречается в популяции ашкенази, составляют около 50% аллелей. Приблизительно 1 из 27 евреев ашкенази — носитель аллеля Тея-Сакса, а встречаемость больных новорожденных в 100 раз выше, чем в других популяциях.
Наиболее вероятным объяснением столь высокой частоты может быть или эффект родоначальника, или селективное преимущество гетерозигот. В этой популяции скрининг носительства облегчен молекулярной характеристикой болезни, поскольку большинство носителей имеет всего один из трех частых аллелей.
Аллели псевдонедостаточности гексозаминидазы А при болезни Тея-Сакса и их клиническое значение
Неожиданное последствие скрининга носительства болезни Тея-Сакса среди популяции евреев ашкенази — открытие уникального класса аллелей, аллелей псевдонедостаточности. Как подразумевает их название, два аллеля псевдонедостаточности клинически благоприятны. Индивидуумы, идентифицированные как псевдонедостаточные при скрининге, — генетические компаунды с аллелем псевдонедостаточности в одной хромосоме и частой мутацией Тея-Сакса в другой.
Эти индивидуумы имеют низкий уровень активности гексозаминидазы А (около 20% в лейкоцитах по сравнению с контролем), но достаточный, чтобы предупредить накопление ганглиозида GM2 в мозге. Значение аллелей псевдонедостаточности гексозаминидазы А двоякое. Во-первых, они усложняют пренатальную диагностику, поскольку плод с псевдонедостаточностью может быть неправильно диагностирован как больной.
Во-вторых, в общем выявление аллеля псевдонедостаточности гексозаминидазы А указывает, что скрининговые программы для других генетических болезней должны учитывать возможность существования схожих аллелей в других локусах, а это может затруднять правильную диагностику при скрининге или диагностических тестах.