Синдром Noonan. Причины и проявления синдрома Ноонан
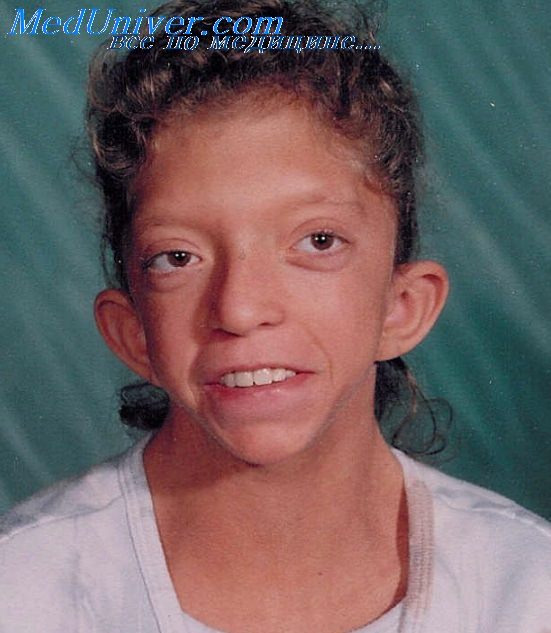
Синдром Noonan плейотропный синдром встречается довольно часто (1 случай на 1000-2500 детей, рожденных живыми) и в основном поражает сердечно-сосудистую систему. Для больных синдромом Noonan, наследуемым по аутосомно-доминантному типу, характерны низкий рост, иногда С необычной деформацией грудины, вальгусная деформация локтевого сустава, складки на шее, врожденная лимфедема и ВПС.
У пациентов с синдромом Noonan отмечают задержку умственного развития, аномалии гемопоэза, которые способствуют развитию лейкемии и крипторхизму. Довольно часто отмечают лимфатическую дисплазию, особенно нижних конечностей, однако клинические осложнения встречаются менее чем в 20% случаев. К наиболее тяжелым поражениям относят хилоторакс (скопление лимфы в плевральной области) и энтеропатию с потерей белка.
Несмотря на фенотипическое сходство синдрома Noonan с синдромом Turner, заболевание поражает и мужчин, и женщин. Синдром Noonan часто называют кардиолицевым синдромом, поскольку для него характерны лицевой дизморфизм (гипертелоризм, птоз) и обширное поражение сердечнососудистой системы (распространенность 80-90%).
Наиболее частым дефектом при синдроме Noonan является стеноз клапанов легочной артерии (диагностируют у 40% больных), который всегда следует подозревать у пациентов с этим синдромом. Створки клапана утолщены и характеризуются дисплазией даже в отсутствие нарушений гемодинамики.
Синдром Noonan - Нунан
Иногда развивается гипоплазия легочной артерии или воронкообразное подклапанное ремоделирование, которое приводит к ГКМП, часто асимметричной, с преобладанием в любом из желудочков. ДМПП встречается примерно у 30% пациентов с синдромом Noonan и обычно ассоциируется со стенозом легочной артерии.
ДМЖП и открытый артериальный проток определяют в 10% случаев. Врожденные аномалии коронарных артерий обычно обнаруживают случайно во время изучения более явных нарушений.
Синдром Noonan обусловлен мутациями двух генов: PTPN11 и KRAS. Большие с синдромом Noonan в 50% случаев имеют мутации в гене PTPN11, который кодирует белок SHP-2 — фермент тирозинфосфатазу, содержащую домен Src-гомолог 2 (SH2), который вовлечен в передачу сигнала с участием RAS-митогенактивируемой протеинкиназы. Связываясь с гуанозиидифосфатом и гуанозинтрифосфатом, RAS-белки обеспечивают регуляцию передачи внутриклеточных сигналов, которые контролируют пролиферацию, дифференциацию и выживаемость клеток.
Мутации гена PTPN11 у человека концентрируются в области тех последовательностей ДНК, которые взаимодействуют с SН2-доменом и доменами, вовлеченными в переключение белка из активного в неактивное состояние; эти мутации приводят к избыточной SН2-активпости и коррелируют со сниженной транскрипционной активацией ядерного фактора активированных Т-клеток. Миссенс-мутации гена KRAS, колирующего 2 изоформы белков семейства RAS, ответственны лишь за 2% случаев синдрома Noonan.
Мутации гена PTPN11, связанного с передачей сигнала через систему RAS-МАПК, и гена KRAS не исключают наличия мутаций в других генах-кандидатах, кодирующих белки этого метаболического пути, которые также могут оказаться причиной развития синдрома Noonan.