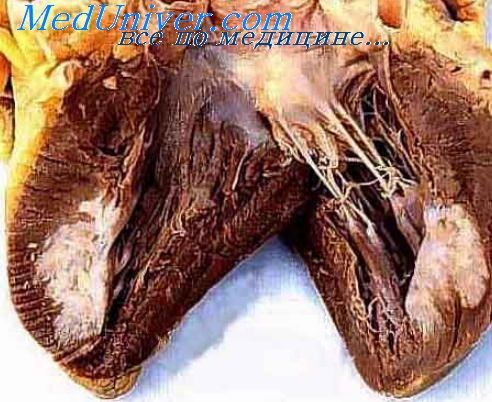
Около 35% случаев первичных опухолей сердца составляют миксомы. Обычно это изолированные доброкачественные опухоли, поражающие левое предсердие (ЛП) у женщин среднего возраста; большинство миксом сердца подлежит хирургическому удалению, они редко рецидивируют, их лечение необходимо для профилактики эмболии и инсультов.
Кардиальные миксомы в 10% случаев являются частью доминантно наследуемого комплекса Carney. В отличие от миксом, возникающих спорадически, опухоли сердца при комплексе Carney поражают мужчин и женщин в равной степени, развиваясь в основном в предсердиях, но затрагивают и желудочки, локализуются во многих участках и, несмотря на полное хирургическое удаление, часто появляются вновь в довольно удаленных друг от друга участках сердца.
К экстракардиальным проявлениям комплекса Carney относят миксомы, лентигиноз, гиперпигментацию и эндокринные нарушения. Клиническая картина заболевания весьма разнообразна даже у членов одной семьи.
Комплекс Carney связан с мутацией гена PRKAR1, кодирующего Rla-регуляторную субъединицу цАМФ-зависимой протеинкиназы А. Все известные мутации этого гена у человека вызывают гаплонедостаточность и снижают активность фермента в 2 раза.
В сердце ген PRKARla может функционировать как ген-супрессор опухоли. Неконтролируемое миксоматозное разрастание может быть следствием как мутации гена PRKAR1а еще на стадии зародыша, так и приобретенной мутации другого гена-супрессора, например в случае мутаций гена NF1 при нейрофиброматозе. При других мутациях, связанных с комплексом Carney, в перинатальном периоде поражаются тяжелые цепи миозин.), что приводит к заболеванию, механизмы развития которого пока не известны.
Наиболее распространенной причиной сердечно-сосудистых заболеваний являются нарушения, наследуемые по законам Менделя. Большинство генов, вызывающих КМП, кодируют структурные белки, участвующие в важнейших процессах мышечного сокращения. Встраиваясь в функциональные комплексы, мутантные структурные белки оказывают разрушительное действие и приводят к развитию заболевания. В противоположность этому мутации, связанные с ВПС, затрагивают факторы транскрипции и сигнальные молекулы, которые регулируют развитие сердца.
При этих мутациях уровень соответствующих белков снижен в 2 раза, что недостаточно для нормального развития сердца во время эмбриогенеза. Мутации, приводящие к развитию ВПС, характеризуются широким спектром клинических проявлений. Значимость клинической диагностики КМП и врожденных заболеваний сердца, основанной на генетическом анализе, все возрастает и позволяет обнаружить заболевание на ранних этапах, а также оценить риск eго развития у членов семьи больного.
Возможности лечения KMП и ВПС пока ограничены. Выявление вовлеченных в их патогенез генов и создание соответствующих экспериментальных моделей, продолжающееся изучение механизмов генетических мутаций позволят увеличить возможности для вмешательства и предотвращения болезней.