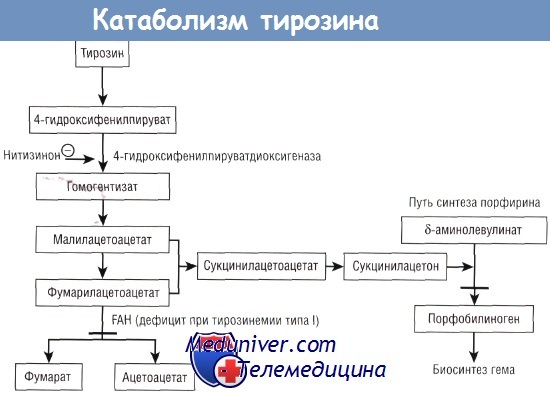
Наследственная тирозинемия представляет собой врожденное нарушение метаболизма тирозина, вызванное дефицитом фермента фумарилацетоацетатгидролазы (FAH), который является последним в пути катаболизма этой аминокислоты. Наиболее часто данное заболевание встречается во «французской» Канаде (Сагеней, Квебек); частота патологии среди новорожденных в этом регионе составляет 1:1846. Уменьшение активности FAH обусловлено изменениями в последовательности нуклеотидов.
У всех новорожденных из района Сагеней и у 28% пациентов из других регионов мира была выявлена сплайсинг-мутация. Большинство этих пациентов были гомозиготны по данной мутации, гуанин был заменен на аденин в нуклеотидной последовательности сплайсинг-донора. Согласно работам других исследователей, ген FAH человека имеет длину 35 килобаз и разбит на 14 экзонов. Отмечены нуклеотидные мутации с заменой тимидина на гуанин и триптофана на стоп-кодон в позиции 262.
Экспрессия FAH в печени пациентов с наследственной тирозинемией была проанализирована на нескольких молекулярных уровнях, включая мРНК и ферментативную активность, приводящих к дифференцировке фенотипических вариантов. У детей с тирозинемией были описаны многие мутации гена FAH; гетерогенность экспрессии FAH в печени этих пациентов отмечена на уровне мРНК, ферментативной и белковой активности.
До недавнего времени не существовало способов терапии детей с наследственной тирозинемией, за исключением раннего назначения диеты с ограничением тирозина и трансплантации печени. Клонирование кодирующей FAH кДНК с помощью трансфера гена ретровируса и интегразы сделало возможным появление в будущем методов генной терапии этого заболевания.
Уже сейчас можно восстановить активность FAH в фибробластах пациентов, имеющих дефицит этого фермента.
Первым задокументированным способом терапии наследственной тирозинемии было применение деривата пестицида (NTBC, или нитизинона [2-(2-нитро-4-трифлуорометилбензол)-1,3-циклогексанедион]). Это химическое соединение нарушает метаболизм тирозина путем ингибирования 4-гидроксифенилпируватдиоксигеназы, препятствуя появлению и накоплению сукцинилацетона и сукцинилацетоацетата. В первых клинических исследованиях пациенты получали перорально нитизинон в дозе 0,1-0,6 мг/кг/сут.
Сукцинилацетон сыворотки быстро снижался до почти неопределяемого уровня, отмечалось улучшение клинического состояния в целом и функции печени, а также уменьшался уровень фетопротеина сыворотки. После первоначального успеха лечения наследственной тирозинемии нитизиноном в Гетеборге (Швеция) было проведено многоцентровое исследование, включившее почти 300 пациентов со всего мира. Многие из этих пациентов получали терапию в течение 5 лет с хорошими результатами. В настоящее время неизвестно, как новые фармакологические препараты будут влиять на возможность развития ГЦК.
Тирозинемия типа I является следствием дефицита фумарилацетоацетат-гидролазы (FAH). Сукцинилацетон ингибирует синтез порфобилиногена. Нитизинон ингибирует фермент 4-гидроксифенилпируватдиоксигеназу.
Поскольку ГЦК гистологически подтверждается у 37% детей с тирозинемией, результаты проведенного в Гетеборге исследования позволяют предположить значительно меньшую частоту этого заболевания у тех пациентов, кому нитизинон назначали до 2-летнего возраста. Рекомендуемая в настоящее время доза нитизинона составляет 1 мг/кг/сут. Препарат применяют на фоне диеты с ограничением фенилаланина и тирозина.
Трансплантацию печени проводят детям с тирозинемией в случае острой печеночной недостаточности, плохого ответа на фармакотерапию и предполагаемой ГЦК. Трансплантация печени решает проблему, связанную как с опухолью печени, так и с метаболическим нарушением наследственной тирозинемии типа I. До начала терапевтического использования нитизинона трансплантация печени рекомендовалась всем детям с наследственной тирозинемией в возрасте до 12 мес.
Несмотря на значительное улучшение метаболического контроля благодаря терапии нитизиноном, ГЦК до сих пор представляет серьезный риск для всех пациентов с наследственной тирозинемией. Нитизинон не корригирует аномальную экспрессию гена и развитие печеночной дисплазии. Риск ГЦК увеличивается при задержке начала терапии нитизиноном, поэтому необходимо его назначать сразу же, как это становится возможным.
Классически наследственная тирозинемия имеет хроническую и острую формы. У пациентов с хронической формой уровень иммунореактивной FAH составляет примерно 20% от нормальной активности фермента. У пациентов с острой формой заболевания не выявляется иммунореактивная FAH на иммуноблотах печени, почек и лимфоцитов. До начала широкого применения нитизинона при наследственной тирозинемии шанс пациента выжить только на диетотерапии был довольно низким.
Выживаемость после появления симптомов заболевания варьирует в зависимости от возраста, в котором возникло заболевание, — чем раньше появились симптомы, тем хуже прогноз. Наиболее частыми причинами смерти являлись печеночная недостаточность и сопутствующее кровотечение (67%), ГЦК (17%) и подобный порфирии синдром с дыхательной недостаточностью (10%). Однолетняя выживаемость с момента начала заболевания у детей в возрасте до 2 мес, между 2 и 6 мес и старше 6 мес составляла 38, 74 и 96% соответственно. На основе этих сроков выживаемости была предложена новая классификация: формы тирозинемии, проявляющиеся очень рано, рано и поздно.
После полученного в январе 2002 г. разрешения от FDA США применять нитизинон отмечается действительное улучшение показателей заболеваемости и смертности от наследственной тирозинемии.
Острая форма наследственной тирозинемии может возникнуть в любом возрасте. Обычно она приводит к тяжелой гепатоцеллюлярной дисфункции и часто сочетается с выраженным повышением уровня фетопротеина в сыворотке. Уровни тирозина в сыворотке также обычно повышены. Диагностическим признаком при данном врожденном нарушении метаболизма тирозина является значительное увеличение содержания сукцинилацетона в моче.
Из 48 детей у 42% была отмечена острая неврологическая патология (криз), не описанная до этого как осложнение. Данное состояние развивалось в среднем в возрасте 1 года и имело следующие симптомы: периферическая нейропатия, сопровождающаяся болью и гипертензией разгибателей; рвота и паралитический илеус; мышечная слабость и попытки членовредительства. Между кризами большинство детей оставались сохранными.
Достоверный биохимический маркер неврологических кризов обнаружен не был, хотя были исследованы уровни тирозина и сукцинилацетона в моче. Экскреция с мочой 8-аминолевулиновой кислоты (нейротоксического промежуточного соединения в биосинтезе порфирина) была повышена во время кризов, но такое же повышение отмечалось и в бессимптомные периоды. Было высказано предположение, что сукцинилацетон может обладать нейротоксическими свойствами. Интересно, что опыты на животных выявили факт, что мозговая ткань секвестрируется сукцинилацетоном и что нарушение биосинтеза тема в мозге может привести к ухудшению окислительного метаболизма. Таким образом, сукцинилацетон может быть ответствен за симптомы поражения ЦНС при тирозинемии.
При хронической форме наследственной тирозинемии можно выявить клубочковую и канальцевую дисфункции, а также вовлечение в патологический процесс печени, нервной и мышечной систем. У детей с длительной почечной дисфункцией может развиться тяжелое интерстициальное заболевание. В эру нитизинона при раннем назначении препарата у большинства пациентов сохраняется нормальная функция почечных канальцев и лишь немногим детям необходима пересадка почек.