Множественная сульфатазная недостаточность у ребенка - кратко с точки зрения педиатрии
Данное АуР-заболевание развивается в результате дефицита ферментной активности по меньшей мере девяти сульфатаз, в т.ч. арилсульфатаз А, В и С и идуронат-2-сульфатазы. Специфический дефект определяется в ферменте (сульфатаз-модифицирующий фактор 1), который входит в С-α-формилглицингенерирующую систему (кодирующий ген располагается в 3p26).
Сульфатаз-модицифирующий фактор 1 ответственен за посттрансляционную модификацию и каталитическую активацию белков семейства сульфатаз. При наличии недостаточности причинного фермента нарушается посттрансляционная модификация всех сульфатаз, чем и объясняется возникновение множественных ферментных нарушений.
На фоне дефицита указанных ферментов в коре больших полушарий ГМ и тканях внутренних органов накапливаются сульфатиды, мукополисахариды, сульфаты стероидов и ганглиозиды. В результате развивается клинический фенотип с признаками лейкодистрофии и мукополисахаридозов. Кроме того, может возникнуть тяжелая форма ихтиоза.
В Саудовской Аравии распространен клинический вариант с симптомами мукополисахаридозов (макроцефалия, множественный дизостоз, помутнение роговицы, сдавление шейного отдела спинного мозга в сочетании с умственной отсталостью).
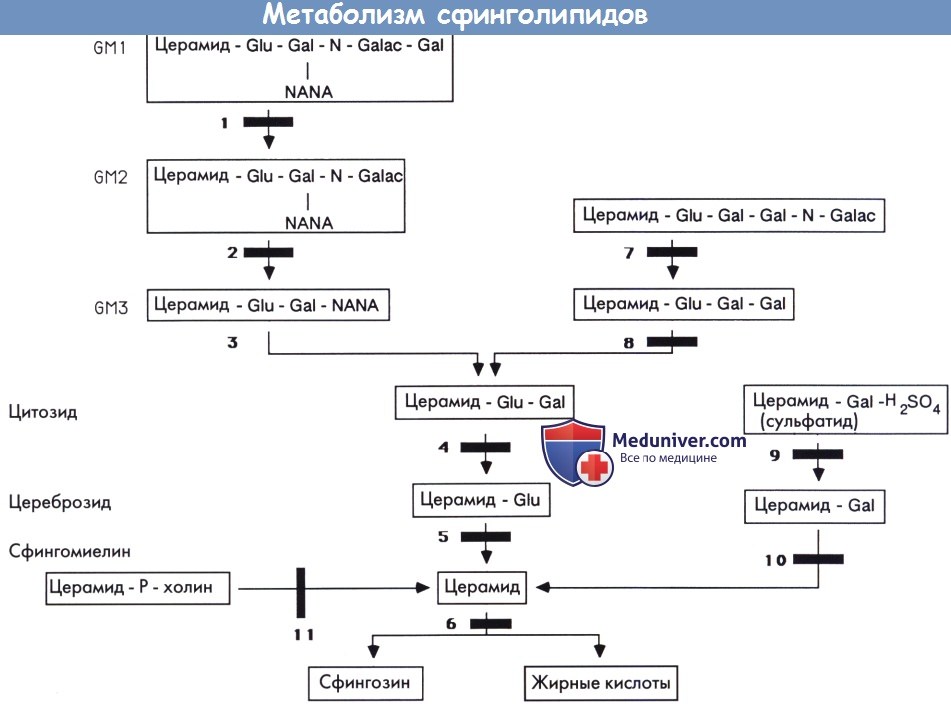
Метаболизм сфинголипидов.
Gal—галактоза. Glu—глюкоза. N-Galac—N-ацетилгалактозамин. NANA—нейраминовая кислота (N-ацетилнейраминовая кислота).
1 — β-галактозидаза. 2 — β-гексозаминидаза А (болезнь Тея-Сакса) и другие ганглиозидозы типа II GМ2.
3 — GM3 сиалидаза. 4 — лактозилцекамидаза (лактозилцерамидоз). 5 — β-глюкозидаза (глюкоцереброзидаза) (болезнь Гоше).
6 — церамидаза (болезнь Фарбера). 7 — β-гексозаминидаза В (болезнь Сандгоффа и другие О варианты GM2 ганглиозидозов).
8 — церамид-тригексозидаза/α-галактозидаза (болезнь Фабри). 9 — цереброзидсульфатаза/арилсульфатаза* (метахроматическая лейкодистрофия).
10 — β-галактозидаза/β-галактоцереброзидаза (болезнь Краббе). 11 — сфингомиелиназа (болезнь Ниманна-Пика А и В).
*Активность, оцененная на искусственных субстратах, не всегда соответствовала активности цереброзиодсульфатазы.
Клиническая картина множественной сульфатазной недостаточности отличается гетерогенностью, возраст дебюта заболевания вариабелен, чаще на втором году жизни. В большинстве случаев заболевание проявляется на 1-2-м году жизни прогрессирующей задержкой психомоторного развития после периода темповой задержки в развитии. На втором году регрессируют приобретенные моторные и речевые навыки, формируется деменция.
У больных развивается спастичность, слепота, тугоухость, нарушения глотания и судороги. Данные проявления характерны для МЛД, у больных также имеют место мягкие, умеренно выраженные проявления мукополисахаридозов («огрубение» черт лица по типу гаргоилизма, гепатоспленомегалия, множественный дизостоз). Ихтиоз развивается на 2-3-м году жизни. Офтальмологические симптомы встречают достаточно редко, среди них атрофия зрительных нервов, пигментная дегенерация сетчатки, симптом «вишневой» косточки, помутнение роговицы.
Летальный исход развивается в конце первого-начале второго десятилетия, в некоторых случаях продолжительность жизни возможна до начала третьего десятилетия*.
P.S. * Захарова Е.Ю., Байдакова Б.Г., Михайлова С.В. и др. Лизосомные болезни накопления: руководство для врачей. М.: ГЭОТАР-Медиа, 2021.
Описан вариант с неонатальной манифестацией у единичных больных, ассоциированный с активностью всех сульфатаз не более 10% от нормы. При таком течении с рождения имеются фациальные дисморфии, гаргоилические черты лица, короткая шея, гепатомегалия, гипоплазия тел позвонков, эпифизарная дисплазия. В дальнейшем — развитие гидроцефалии, ихтиоза и помутнения роговицы.
Обследование на носительство и пренатальную диагностику можно проводить путем определения ферментной активности или специфических генных нарушений. Др. методов специфической терапии, кроме поддерживающей, при множественной сульфатазной недостаточности не существует.