Болезни Вольмана и накопления эфиров холестерина (ХС) относятся к АуР-лизосомным болезням накопления, их проявления обусловлены дефицитом лизосомной кислой липазы (ДЛКЛ) и накоплением эфиров ХС и триглицеридов в гистиоцитарных клетках большинства внутренних органов; нагруженные липидами клетки приобретают характерный «пенистый» вид. ДЛКЛ обусловлен мутациями гена LAL (LIPA)*, который картирован на хромосоме 10 (10q24-q25).
P.S. * Ген LAL в КР назван LIPA, это название является синонимом, в тексте выполнена замена.
В международной базе данных по мутациям человека описано ок. 100 мутаций в гене LIPA. При тяжелых формах ДЛКЛ заболевания выявляются нонсенс-мутации, крупные перестройки гена, мутации со сдвигом рамки считывания, в гомозиготном или компаундгетерозиготном состоянии. Наиболее распространенный патогенный аллель — синонимичная замена, нарушающая сайт сплайсинга в экзоне 8 c.894G>A (E8SJM-1G>A), является причиной заболевания в >50% известных случаев.
Накопление эфиров ХС и триглицеридов в органах и тканях сопровождается дислипидемией, которая характеризуется повышением в крови уровня ХС, ЛПНП, а уровень ЛПВП соответствует норме или снижен. У некоторых больных имеет место гипертриглицеридемия.
Распространенность ДЛКЛ варьирует в зависимости от факторов этнической принадлежности и географического положения. Частота ДЛКЛ составляет в среднем 1:40 000-1:300 000 живых новорожденных. Частота встречаемости младенческой формы заболевания болезни Вольмана составляет 1:100 000-500 000 живорожденных новорожденных. Исследования по изучению частоты встречаемости ДЛКЛ в РФ проведены в г. Москва, получены данные частоты приблизительно 1:70 000 новорожденных.
Выделяют две основные формы ДЛКЛ:
• инфантильная (болезнь Вольмана) — с манифестацией в младенческом возрасте;
• болезнь накопления эфиров холестерина (БНЭХ) — с дебютом в возрасте старше 1 года (наиболее часто в 2-5 лет).
Проявления ДЛКЛ представляют собой спектр клинических фенотипов с вариабельным характером течения и прогнозом болезни.
При инфантильной форме ДЛКЛ — болезни Вольмана активность ЛКЛ составляет <1% от нормы, что приводит к быстрому массивному накоплению эфиров ХС и триглицеридов в лизосомах органов и тканей, прежде всего в печени, селезенке, надпочечниках, ворсинах кишечника, костном мозге, лимфатических узлах, в макрофагах РЭС, что определяет полисистемный характер течения болезни.
P.S. * КР «Другие нарушения накопления липидов (Дефицит лизосомной кислой липазы)», возрастная группа: дети/взрослые, год утверждения — 2020. Разработчик клинической рекомендации: Ассоциация медицинских генетиков, Союз педиатров России, Российская гастроэнтерологическая ассоциация.
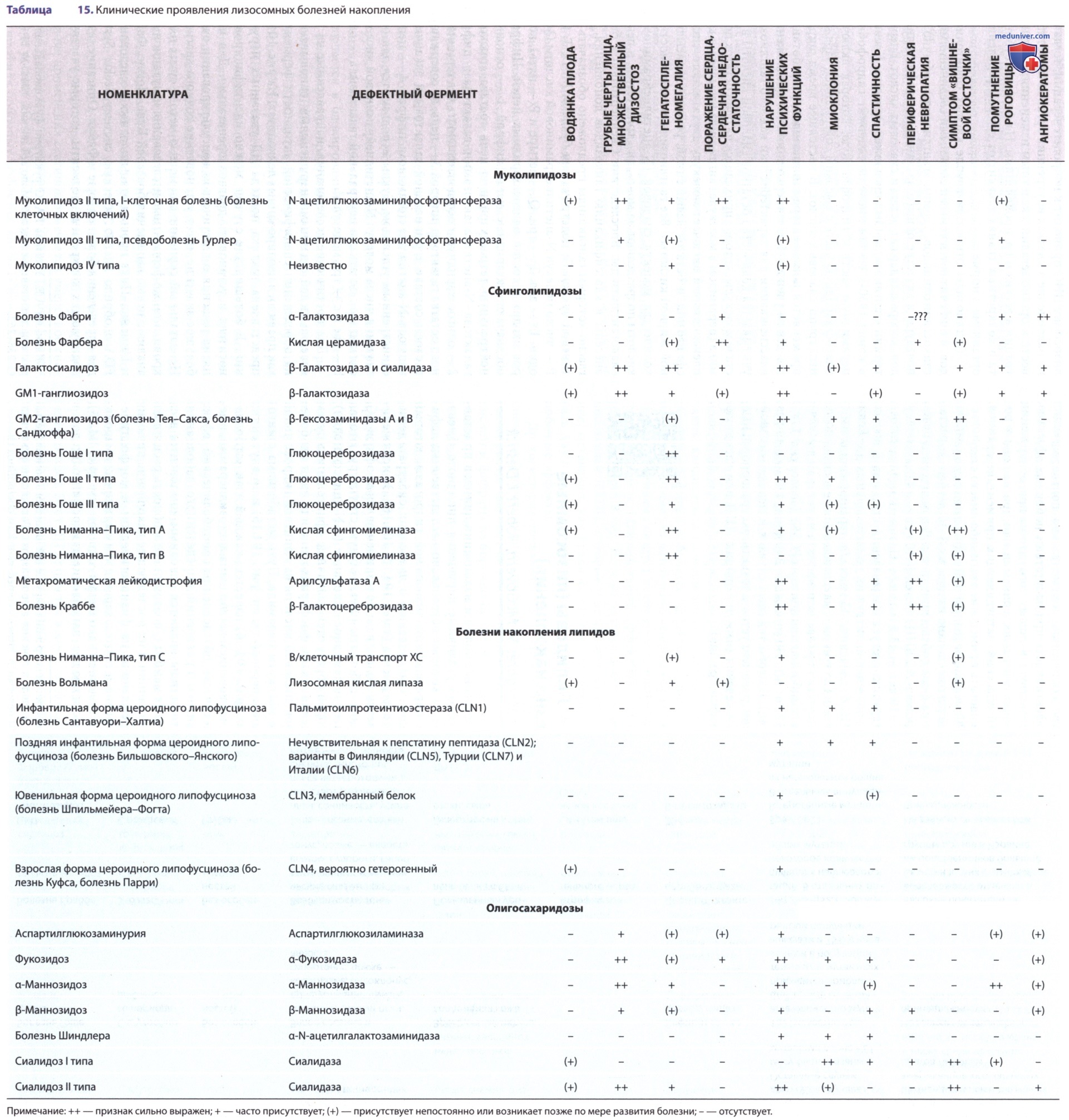
Болезнь Вольмана отличается наиболее тяжелым клиническим фенотипом и характеризуется фатальным метаболическим расстройством грудного возраста. Клинические симптомы проявляются на первой неделе жизни и включают задержку роста, неукротимую рвоту, вздутие живота, стеаторею и гепатоспленомегалию (см. табл. 15).
Возможно развитие симптомов в течение первых месяцев жизни. Часто отмечается увеличение объема живота за счет вздутия кишечника, гепатомегалии и/или ге-патоспленомегалии, снижение МТ вплоть до кахексии в тяжелых случаях. Также у этих пациентов развиваются интермиттирующая лихорадка, вялость, астения и гиперрефлексия, анемия и тромбоцитопения.
Как правило, развивается гиперлипидемия. Могут возникнуть нарушения функции и фиброз печени. У 50% пациентов наблюдают кальцификацию надпочечников.
По данным визуализирующих методов, увеличенные надпочечники сохраняют свою полулунную или пирамидальную форму, по всей паренхиме их определяются точечные очаги кальцификации. Отсутствие кальцификатов надпочечников не исключает диагноз. Больным с болезнью Вольмана присуща неврологическая симптоматика, обусловленная печеночной энцефалопатией. Развитие синдрома мальабсорбции объясняется недостатком жирных кислот, повреждением слизистой оболочки кишечника.
В биохимическом анализе крови — выраженное повышение уровня трансаминаз, значительное увеличение билирубина, синдром холестаза. Частым лабораторным признаком при болезни Вольмана является повышение уровней лактатдегидрогеназы и сывороточного ферритина на фоне снижения Hb, а также признаки гипокоагуляции. При исследовании липидного профиля выявляется дислипидемия: повышение общего ХС, описанное у 81-90% пациентов, за счет ЛПНП (в 95% случаев); снижение ЛПВП — у 85-89% пациентов; повышение триглицеридов — у 48% пациентов.
Известны случаи манифестации болезни Вольмана во в/утробном периоде в виде некроза надпочечников, полигидрамниона, накопления эфиров ХС в органах и тканях и микровезикулярного стеатоза печени. Болезнь Вольмана м.б. причиной в/утробной гибели плода.
Для точной морфологической диагностики в настоящее время разработана иммуногистохимическая панель АТл. Обнаружение катепсина D и экспрессия мембранных лизосомальных маркеров [лизосомально-ассоциированные мембранные белки 1 и 2 (LAMP 1 и 2; англ. Lysosomal-associated membrane protein 1), лизосомальный интегральный мембранный белок 2 (LIMP2; англ. Lysosomal integral membrane protein 2} вокруг липидных вакуолей подтверждает в/лизосомальное накопление липидов. К гист. особенностям относят также накопление цероида в макрофагах.
Летальный исход обычно развивается в течение первых 6 мес жизни.
Болезнь накопления эфиров холестерина протекает менее тяжело, чем болезнь Вольмана, и может оставаться недиагностированной до взрослого возраста.
БНЭХ характеризуется наличием у больных остаточной активности ЛКЛ in vitro в диапазоне 1-12% от нормы.
Гепатомегалия м.б. единственным обнаруживаемым симптомом, хотя больные с этим заболеванием входят в группу высокого риска раннего развития цирроза печени и атеросклероза. У пациентов с ранним тяжелым манифестным течением заболевания может развиваться кальцификация надпочечников.
В РФ кальцификация надпочечников описана лишь у 5% пациентов с БНЭХ.
Заболевание дебютирует в возрасте старше одного года. БНЭХ характеризуется медленным прогрессированием и, как правило, отсутствием неврологической симптоматики. Т.к. ДЛКЛ является мультисистемным заболеванием, симптомы могут быть обусловлены поражением разл. органов, основными мишенями являются печень, селезенка, ЖКТ, почки, сосуды. Медиана появления первых клинических симптомов заболевания составляет 5 лет, но возможна манифестация и во взрослом возрасте.
БНЭХ проявляется гепатомегалией (больше за счет правой доли печени), далее формируется фиброз и цирроз печени с развитием печеночной недостаточности. Синдром цитолиза (увеличение уровней ACT и АЛТ) наблюдается практически у всех пациентов и считается одним из первых проявлений заболевания. Активность этих ферментов в крови варьирует в широком диапазоне (от 2 до 5 норм до превышения данных показателей в 10-20 раз. Спленомегалия обнаруживается у 74% больных, появляются признаки гиперспленизма (анемия, лейкопения, тромбоцитопения). При прогрессировании болезни развивается фиброз и цирроз печени, проявляющийся гепатоспленомегалией, желтухой, асцитом, варикозным расширением вен пищевода.
Дислипидемия проявляется повышением в сыворотке крови уровня общего ХС, ХС-ЛПНП и триглицеридов при нормальном или низком уровне ХС-ЛПВП (гиперлипопротеинемия IIb-типа), что является фактором риска ускоренного развития атеросклероза. Гиперхолестеринемия определяется у 81% пациентов с БНЭХ, увеличение концентрации ХС-ЛПНП имеет место у всех пациентов. На фоне терапии статинами гиперхолестеринемия сохраняется у четверти, а высокий уровень ХС-ЛПНП — у половины больных.
При анализе аполипопротеинограммы у большинства пациентов выявляют повышение уровня основного аполипопротеина ЛПНП — апоВ. Из-за вовлечения в патологический процесс кишечника у ряда больных наблюдаются диарея, боли в животе, стеаторея разной степени выраженности. Наличие БНЭХ можно считать вероятным диагнозом у молодых пациентов с гепатомегалией, криптогенным циррозом печени, увеличением АЛТ, ACT в сыворотке крови, гиперхолестеринемией, гипертриглицеридемией, повышением ЛПНП и неполным ответом на терапию статинами.
а) Диагностика. Физикальная диагностика болезни Вольмана — основные клинические проявления:
• увеличение размеров печени;
• увеличение размеров селезенки;
• увеличение размеров живота;
• синдром мальабсорбции;
• гипотрофия, вплоть до грубой задержки физического развития;
• прогрессирующая задержка психомоторного развития, гиперрефлексия;
• желтушность кожных покровов;
• признаки СН.
Физикальная диагностика болезни накопления эфиров холестерина — основные клинические проявления:
• увеличение размеров печени;
• увеличение размеров селезенки.
б) Лабораторные диагностические исследования:
1. Лабораторные диагностические исследования при инфантильной форме дефицита лизосомной кислой липазы. Всем пациентам с подозрением на инфантильную форму ДЛКЛ (болезнь Вольмана) для верификации диагноза рекомендовано определение активности лизосомной кислой липазы в пятнах крови, высушенных на фильтровальной бумаге. Определение активности ЛКЛ в сухих пятнах крови является «золотым стандартом» диагностики ДЛКЛ. Пациентам, у которых выявлено снижение активности ЛКЛ в пятне крови, рекомендуется проведение молекулярно-генетического исследования (выявление мутаций в гене LIPA).
Всем пациентам с подозрением на инфантильную форму ДЛКЛ (болезнь Вольмана) для выявления анемии, лейкопении, тромбоцитопении рекомендуется проведение развернутого ОАК. Всем пациентам с болезнью Вольмана также проводят биохим. анализ крови всем пациентам с болезнью Вольмана при диагностике и в процессе динамического наблюдения для определения функционального состояния печени: АЛТ, ACT, общий и прямой билирубин, ХС, ХС-ЛПНП, ХС-ЛПВП, триглицериды, гаммаглутамилтрансфераза, ЩФ, альбумин. Частота исследования биохим. анализа крови определяется состоянием пациента.
При диагностике оцениваются АЛТ, ACT, общий и прямой билирубин, общий ХС, ХС-ЛПНП, ХС-ЛПВП, триглицериды. В динамике оцениваются АЛТ, ACT, общий и прямой билирубин, общий ХС (не менее 1 раза в неделю), ЛПНП, ЛПВП, триглицериды (не менее 1 раза в месяц). Детям первого года жизни с подозрением на болезнь Вольмана рекомендовано определение уровня ЛДГ, ферритина и СРВ.
Всем пациентам с болезнью Вольмана при диагностике и в динамике необходимо исследование коагулограммы с целью контроля синтетической функции печени и своевременного предупреждения осложнений, связанных с нарушением гемостаза: АЧТВ, тромбиновое время, фибриноген, ПТВ в крови или в плазме, МНО.
Пациентам с болезнью Вольмана рекомендована лабораторная оценка функции надпочечников с целью своевременной диагностики надпочечниковой недостаточности и назначения заместительной терапии; при выявлении признаков кальцификации надпочечников: исследование уровня общего кортизола в крови, уровня АКТГ в крови, уровня глюкозы в крови, определение рениновой активности плазмы крови, а также содержание натрия и калия в крови.
2. Лабораторные диагностические исследования при болезни накопления эфиров холестерина. Всем пациентам с подозрением на БНЭХ первоначально определяется активность ЛКЛ в пятнах крови, при снижении активности фермента проводится молекулярно-генетическое исследование. Также рекомендуется проведение развернутого ОАК всем пациентам с подозрением на БНЭХ и далее в динамике, в среднем не реже 1 раза в 6 мес с целью выявления анемии, лейкопении, тромбоцитопении.
Всем пациентам с БНЭХ при диагностике и в ходе динамического наблюдения рекомендован биохимический анализ крови в среднем каждые 3-6 мес. К рекомендуемым по исследованию показателям относятся: АЛТ, ACT, общий и прямой билирубин, ХС, ХС-ЛПНП, ХС-ЛПВП, триглицериды, гаммаглутамилтрансфераза, ЩФ, альбумин. Всем пациентам с БНЭХ при диагностике и в динамике рекомендовано исследование коагулограммы по следующим показателям: АЧТВ, тромбиновое время, фибриноген, ПТВ в крови или в плазме, МНО.
Всем пациентам с БНЭХ также проводят исследование уровня мочевины, креатинина в сыворотке крови с определением СКФ при диагностике заболевания и при динамическом наблюдении, в среднем каждые 6-12 мес с целью контроля функции почек и своевременной диагностики ее нарушения.
в) Инструментальные диагностические исследования:
1. Инструментальные диагностические исследования при инфантильной форме дефицита лизосомной кислой липазы (болезни Вольмана). Всем пациентам с подозрением на болезнь Вольмана рекомендовано проведение комплексного УЗИ брюшной полости и забрюшинного пространства (в т.ч. УЗИ надпочечников), УЗДС сосудов печени (УЗДС сосудов гепатобилиарной зоны), УЗДС сосудов селезенки и после лабораторного подтверждения диагноза — с целью контроля состояния внутренних органов.
Частота проведения комплексного УЗИ органов БП и забрюшинного пространства (в т.ч. УЗИ надпочечников) диктуется состоянием пациента, но не менее 1 раза в месяц.
Пациентам с болезнью Вольмана рекомендовано проведение ФЭГДС после лабораторного подтверждения диагноза при наличии признаков портальной гипертензии, ультразвуковых признаков нарастания размеров селезенки и расширения воротной и селезеночной вен. Предпочтительно использование неинвазивных методик, напр. эластографии печени (эластометрии печени), МРТ органов БП.
2. Инструментальные диагностические исследования при болезни накопления эфиров холестерина с дебютом в возрасте старше 1 года. Всем пациентам с подозрением на БНЭХ рекомендовано проведение комплексного УЗИ БП и забрюшинного пространства, а также УЗДС сосудов печени (УЗДС сосудов гепатобилиарной зоны), УЗДС сосудов селезенки и после лабораторного подтверждения диагноза — с целью динамического контроля состояния внутренних органов. УЗИ забрюшинного пространства, в т.ч. УЗИ надпочечников, может выявить кальцификаты надпочечников, но у пациентов с БНЭХ кальцификация надпочечников встречается не более чем в 5% случаев.
Пациентам с БНЭХ после лабораторного подтверждения диагноза рекомендовано проведение МРТ органов БП и/или эластографии печени (эластометрии печени) с целью контроля состояния печени при проведении ФЗТ. Частота исследований в среднем: МРТ органов брюшной полости — 1 раз в год, эластография печени — 1 раз в 6 мес.
Пациентам с БНЭХ рекомендовано проведение ФЭГДС после лабораторного подтверждения диагноза при наличии признаков портальной гипертензии, ультразвуковых признаков нарастания размеров селезенки и расширения воротной и селезеночной вен.
Для оценки состояния ССС рекомендовано ЭКГ, ЭхоКГ в среднем каждые 1-2 года при наличии атеросклероза и каждые 2-5 лет — у стабильных пациентов. При наличии аритмии необходимо проведение суточного мониторирования ЭКГ, по показаниям — стресс-теста в соответствии с рекомендациями по ведению пациентов с атеросклерозом и аритмией.
Всем пациентам с установленным диагнозом БНЭХ рекомендовано проведение ультразвукового допплерографического анализа сосудов головы и шеи: УЗИ брахиоцефальных структур для диагностики атеросклероза и оценки степени его выраженности. Дополнительно м.б. проведено определение лодыжечно-плечевого (лодыжечно-брахиального) индекса, исследование кальцификации коронарных артерий.
Взрослым пациентам с БНЭХ рекомендовано проведение при рекомендации кардиолога в соответствии с общими принципами подхода к диагностике атеросклероза компьютерно-томографической ангиографии сосудов ГМ и компьютерно-томографической ангиографии брахиоцефальных артерий для оценки состояния сердечнососудистой системы. Для подтверждения диагноза БНЭХ не рекомендована биопсия печени в качестве рутинного метода.
г) Основные принципы диагностики и ведения больных:
1. Диагностика и ведение больных с инфантильной формой дефицита лизосомной кислой липазы (болезнь Вольмана). Пациентов с болезнью Вольмана наблюдает врач-гастроэнтеролог (и/или врач-педиатр), врач-генетик, показана консультация врача-диетолога с целью назначения низкожировой диеты, коррекции мальабсорбции (подбор специализированной диеты с ограничением животных жиров, а в ряде ситуаций и парентерального питания), детского кардиолога, врача-невролога, врача-трансплантолога при наличии показаний для проведения трансплантации печени. В жизнеугрожающих состояниях необходимо участие врача-анестезиолога-реаниматолога.
Пациентам с установленным диагнозом «болезнь Вольмана» и наличием изменений в надпочечниках рекомендована консультация врача-эндокринолога с целью оценки функции надпочечников и своевременного назначения гормонозаместительной терапии.
2. Диагностика и ведение больных с болезнью накопления эфиров холестерина. Пациентов с БНЭХ наблюдает врач-гастроэнтеролог, врач-терапевт (в детской практике — врач-педиатр), а также врач-генетик, показана консультация врача-диетолога с целью назначения низкожировой диеты (подбор специализированной диеты с ограничением животных жиров), врача-кардиолога (оценка сосудистого риска, назначение лечения первично и в последующем каждые 6 мес), врача-трансплантолога при наличии показаний для трансплантации печени. При наличии неврологических нарушений: консультация врача-невролога (первично и далее ежегодно).
Установление диагноза и выявление носителей основаны на определении активности ЛКЛ в лейкоцитах периферической крови или культуре кожных фибробластов. В гене LIPA обнаружены разл. мутации, вызывающие заболевание.
В настоящее время гено-фенотипическая корреляция установлена только на уровне клинической формы ДЛКЛ (болезнь Вольмана либо БНЭХ), но не прослеживается для отдельных фенотипических признаков, молекулярно-генетическое исследование применяется для подтверждения, а не установления диагноза ДЛКЛ. Выявление семейной мутации гена LIPA делает возможным обследование родственников пробанда, а также проведение пренатальной и преимплантационной диагностики.
Для любой последующей беременности в семьях, отягощенных хотя бы одним случаем ДЛКЛ (болезнь Вольмана, БНЭХ), рекомендуется проведение пренатальной диагностики, но в случае легких форм болезни решение о ее проведении должно быть принято после подробного обсуждения с семьей вероятных рисков.
Пренатальная диагностика основывается на определении пониженных уровней ЛКЛ или обнаружении специфических мутаций в культуре клеток ворсинок хориона или амниоцитов.
д) Лечение. Для лечения применяют ЛП, подавляющие синтез ХС, в сочетании с холестирамином и коррекцией диеты, однако клиническая эффективность терапии невелика или практически отсутствует.
Лечение ДЛКЛ включает патогенетическую терапию — назначение ФЗТ и симптоматическое лечение.
1. Патогенетическое лечение при инфантильной форме ДЛКЛ (болезни Вольмана). Всем пациентам с установленным диагнозом «болезнь Вольмана» рекомендовано проведение ФЗТ.
Себелипаза альфа — рекомбинантная форма ЛКЛ, одобренная к применению FDA в 2015 г. По данным клинического исследования, 67% детей грудного возраста с дефицитом ЛКЛ, получавших терапию этим ЛП, дожили до возраста >12 мес в сравнении с 0% детей из катамнестической группы сравнения, в которой пациенты не получали лечения себелипазой альфа и все умерли к восьми месяцам.
Рекомендованный режим дозирования препарата себелипаза альфа («Канума») зависит от возраста. Детям до 6 мес рекомендуемая начальная доза составляет 1 мг/кг, в последующем — 3 мг/кг в/в 1 раз в неделю, введение ЛП осуществляется парентерально в виде в/в инфузий. В зависимости от клинического ответа следует рассмотреть возможность постепенного увеличения дозировки до 5 мг/кг 1 р/нед. Именно эти дозы позволяют быстро купировать тяжелые осложнения заболевания. Необходим мониторинг основных параметров течения заболевания в динамике ФЗТ.
У пациентов с болезнью Вольмана, получающих ФЗТ при в/в введении себелипазы альфа, могут развиться побочные реакции (лихорадка, озноб, рвота, крапивница, тяжелые реакции гиперчувствительности аллергического типа), происходящие во время инфузии или в течение дня после проведения инфузии. При появлении побочных реакций во время/после инфузии рекомендуется соответствующее лечение, при котором необходимо следовать современным рекомендациям.
2. Симптоматическая терапия при инфантильной форме дефицита лизосомной кислой липазы (болезни Вольмана). При поражении печени при наличии синдромов цитолиза и холестаза рекомендовано назначение ЛП с гепатопротективным действием: препаратов урсодезоксихолевой кислоты. Рекомендовано назначение поливитаминных ЛП в связи с недостаточным их усвоением из пищи, обусловленным нарушением кишечного всасывания у части пациентов с болезнью Вольмана.
Пациентам с болезнью Вольмана и надпочечниковой недостаточностью рекомендовано проведение заместительной терапии минералокортикоидами и/или ГКС согласно соответствующим рекомендациям.
3. Патогенетическое лечение при болезни накопления эфиров холестерина (БНЭХ) с дебютом. По данным исследования с участием 66 детей и взрослых с болезнью накопления эфиров ХС, у пациентов, получавших себелипазу альфа («Канума»), наблюдали значительное снижение уровней АЛТ сыворотки и уменьшение количества жировых отложений в печени, а также улучшение показателей ХС-ЛПНП, триглицеридов и ХС-ЛПВП в сравнении с показателями пациентов, получавших плацебо.
Пациентам с установленным диагнозом БНЭХ рекомендовано проведение ФЗТ с целью замедления прогрессирования заболевания, уменьшения размеров печени и селезенки, регресса или стабилизации фиброза печени, устранения дислипидемии, уменьшения накопления эфиров ХС в органах и тканях.
ЛП для ФЗТ является себелипаза альфа ЛП вводится в виде в/винфузий. Детям >6 мес и взрослым ЛП вводят из расчета 1 мг/кг каждые 2 нед. Необходимо соблюдать интервалы между инфузиями, недопустимы перерывы в терапии, т.к. нарушение режима лечения сопровождается потенциальным риском ухудшения состояния пациента и прогрессирования симптомов. Необходим мониторинг основных параметров течения заболевания.
При обследовании сиблингов (братьев и сестер пробанда) м.б. выявлены дети с ДЛКЛ, не имеющие клинических проявлений. Такие пациенты нуждаются в наблюдении, начинать их лечение необходимо при появлении первых симптомов болезни.
4. Симптоматическое лечение при болезни накопления эфиров холестерина. Пациентам с БНЭХ рекомендован рацион со сниженным содержанием жира. В РФ приняты методические рекомендации МР 2.3.1.2432-08 «Нормы физиологических потребностей в энергии и пищевых в-вах для различных групп населения Российской Федерации». Допускается ограничение в рационе жиров до 30% от энергетической ценности суточного рациона. Доли насыщенных, моно- и полинасыщенных жиров должны быть равными. Максимально допустимое количество ХС — 300 мг/сут, возможно его снижение до 200 мг/сут.
Уровень белка должен соответствовать физиол. норме потребления в зависимости от возраста с увеличением доли растительных белков. Рекомендуемое соотношение растительных и животных белков соответствует 1:1. Доля углеводов должна составлять 50-60% от энергетической ценности рациона. Из них 7-10% — легкоусвояемые.
При заболеваниях печени при наличии синдрома цитолиза и холестаза рекомендовано назначение препаратов с гепатопротективным действием: гепатопротекторное средство растительного происхождения (расторопши пятнистой. экстракт), тиоктовая кислота, урсодезоксихолевая кислота. Рекомендовано назначение поливитаминных ЛП в связи с недостаточным их усвоением, обусловленным нарушением кишечного всасывания части пациентов с ДЛКЛ.
5. Хирургическое лечение:
- Хирургическое лечение при инфантильной форме ДЛКЛ (болезни Вольмана). Пациентам с болезнью Вольмана на стадии декомпенсированного цирроза печени при неэффективности проведения патогенетической терапии рекомендовано рассмотреть вопрос о проведении трансплантации печени. Трансплантация печени ассоциирована с высоким риском осложнений и не оказывает влияния на системные проявления заболевания, не останавливает прогрессию болезни Вольмана и поражение др. органов-мишеней. Сохраняется риск повреждения трансплантированной печени, т.к. уровень ЛКЛ по-прежнему остается низким.
Пациентам с болезнью Вольмана не рекомендовано проведение трансплантации гемопоэтических стволовых клеток в качестве первой линии терапии. В настоящее время недостаточно данных о преимуществах и недостатках данного вида процедуры, также она ассоциирована с высоким риском развития осложнений (отторжение трансплантата, реакция «трансплантат против хозяина» и др. постоперационные осложнения). Поэтому трансплантация гемопоэтических стволовых клеток не рекомендована как рутинная терапевтическая опция для пациентов с болезнью Вольмана.
- Хирургическое лечение при болезни накопления эфиров холестерина с дебютом в возрасте старше 1 года. Пациентам с БНЭХ на стадии декомпенсированного цирроза печени при невозможности или неэффективности проведения патогенетической терапии рекомендуется рассмотреть вопрос о проведении трансплантации печени.
Для пациентов с БНЭХ, основной причиной смертности которых является цирроз и печеночная недостаточность, трансплантация печени была признана успешной тактикой лечения. Имеются публикации, подтверждающие многолетнюю ремиссию пациентов после операции с восстановлением нормального роста, развития и функции печени.
6. Медицинская реабилитация, профилактика и диспансерное наблюдение больных с дефицитом лизосомной кислой липазы. Пациентам с ДЛКЛ (болезнь Вольмана, БНЭХ) и членам их семей рекомендуются консультации психолога/медицинского психолога. Поскольку заболевание носит прогрессирующий пожизненный характер, пациенту необходима помощь, чтобы «принять» диагноз, адаптировать его к жизни с тем, чтобы можно было максимально реализовать его способности к обучению и самостоятельной жизни в дальнейшем.
После установления диагноза ДЛКЛ пациенту и/или его официальным представителям также рекомендуется консультация врача-генетика с целью разъяснений генетического риска, обсуждения возможностей пренатальной и преимплантационной диагностики.
Семьям с детьми с ДЛКЛ рекомендуется медикогенетическое консультирование с целью определения генетического риска. Как и при др. АуР-заболеваниях, при ДЛКЛ для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть ребенок с ДЛКЛ, возможно проведение пренатальной и преимплантационной диагностики. Родителей направляют в специализированные диагностические лаборатории и медцентры.