I-клеточная болезнь [муколипидоз типа II (МЛ II)] и псевдополидистрофия Гурлер [муколипидоз типа III (МЛ III)] — редкие АуР-расстройства, которые имеют некоторые клинические признаки синдрома Гурлер (см. главу 107). Эти заболевания возникают в результате аномального накопления вновь синтезированных лизосомальных ферментов, которые обычно имеют фосфорилированные остатки маннозы для связывания с маннозо-6-фосфатными рецепторами, которые транспортируют ферменты к лизосомам.
Такие остатки маннозо-6-фосфата синтезируются в аппарате Гольджи в ходе двухэтапной реакции при участии двух ферментов. Дефекты лизосомного фермента N-ацетилглюкозамин-1-фосфотрансферазы, катализирующего первый этап, определяются как при МЛ II, так и при муколипидозе III. Оба заболевания представляют собой аллельные нарушения, возникающие в результате мутаций в гене, кодирующем предшественник α/β-субъединиц N-ацетилглюкозамин (GlcNAc)-фосфотрансферазы (GNPTAB).
Дефицит этого фермента приводит к нарушению накопления лизосомных ферментов, в результате чего они секретируются во внеклеточный матрикс. Поскольку для функционирования лизосомных ферментов требуется кислая среда лизосом, у пациентов с указанным нарушением вследствие в/клеточного дефицита большинства лизосомных ферментов накапливаются разл. субстраты. Диагноз МЛ II и МЛ III можно установить по повышенной активности лизосомных ферментов в сыворотке крови или по сниженной активности ферментов в культуре кожных фибробластов. Кроме того, можно определить активность фосфотрансферазы напрямую.
При обоих нарушениях диагноз можно установить пренатально путем определения активности лизосомных ферментов в амниоцитах или клетках ворсинок хориона. Носителей при обоих нарушениях можно выявить путем определения активности ферментов в культуре кожных фибробластов или методом мутационного анализа причинного гена. I-клеточную болезнь можно диагностировать по результатам неонатального скрининга методом тандемной масс-спектрометрии.
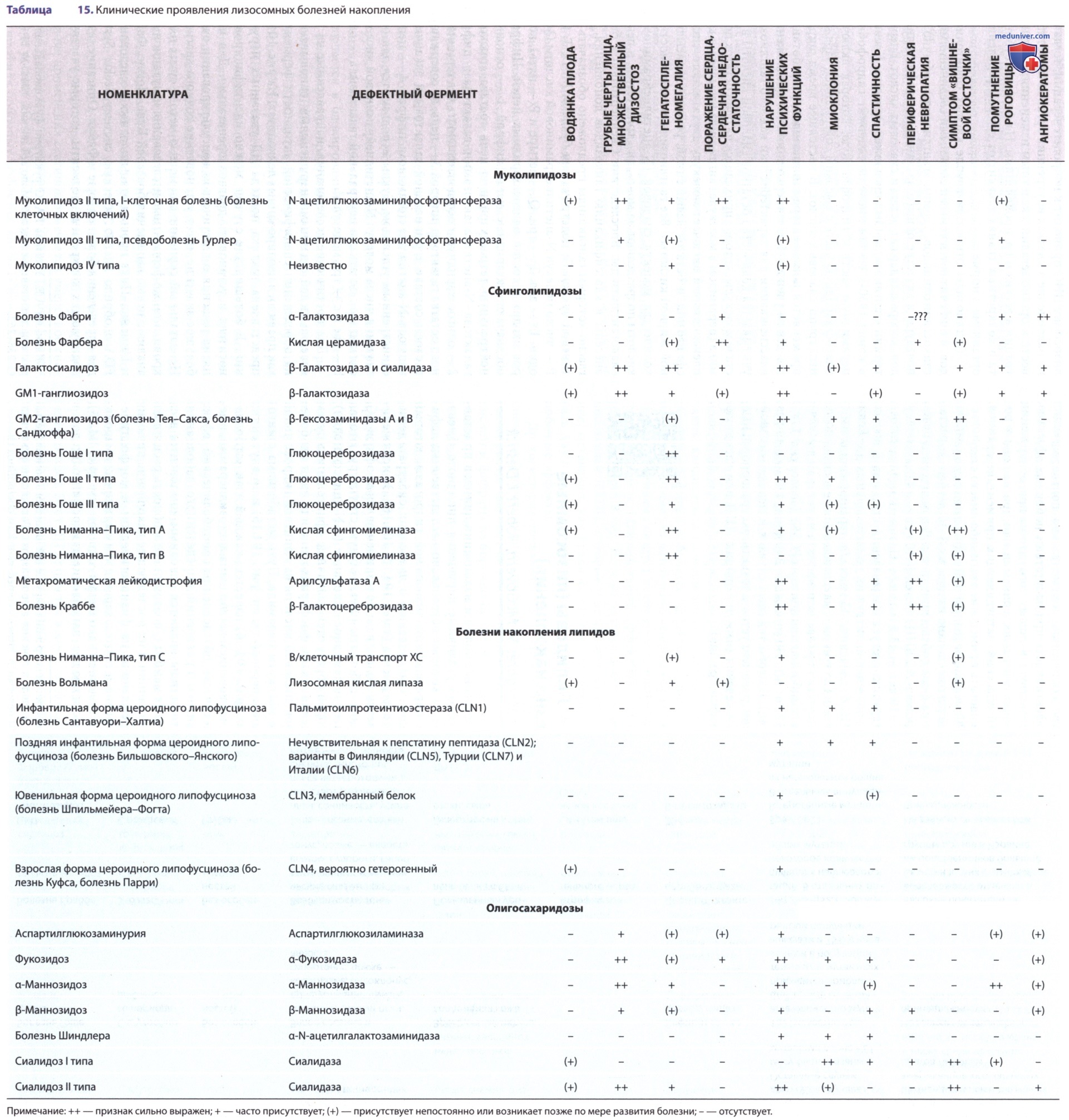
а) I-клеточная болезнь. I-клеточная болезнь, или МЛ II, имеет много общих клинических проявлений с синдромом Гурлер, однако для нее не характерна мукополисахаридурия, а симптомы появляются в более раннем возрасте (см. табл. 15). У некоторых пациентов клинические признаки заболевания (грубые черты лица, черепно-лицевые аномалии, ограничение объема движений в суставах и мышечная гипотония) появляются с рождения.
У плода м.б. неиммунная водянка. У остальных пациентов симптомы проявляются на первом году жизни в виде тяжелой задержки психомоторного развития, грубых черт лица, а также проявлений со стороны костной системы, в т.ч. кифосколиоза и поясничного горба. Кроме того, у пациентов м.б. врожденный вывих ТБС, паховые грыжи и гипертрофия десен. Тяжелые прогрессирующие психомоторные нарушения приводят к летальному исходу в раннем детском возрасте. Лечения 1-клеточной болезни не существует.
б) Псевдополидистрофия Гурлер. Псевдополидистрофия Гурлер, или МЛ III, представляет собой менее тяжелое расстройство, чем I-клеточная болезнь. Первые симптомы появляются в более позднем возрасте, зарегистрированы случаи доживания до взрослого возраста. Симптомы у детей, страдающих этим заболеванием, могут появляться в возрасте ок. 4-5 лет в виде ригидности суставов и низкого роста. Имеются признаки прогрессирующего разрушения тазобедренных суставов и умеренного множественного дизостоза. К характерным рентгенологическим признакам относят низкие крылья подвздошных костей, уплощение проксимальных эпифизов бедренных костей и вальгусную деформацию шейки бедренной кости, а также гипоплазию передней трети поясничных позвонков.
Офтальмологические симптомы включают помутнение роговицы, ретинопатию и астигматизм. Жалобы на нарушение зрения отмечают редко (см. табл. 15). У некоторых пациентов наблюдают нарушение способности к обучению и интеллектуальные нарушения. Лечение, которое должно включать ортопедическую помощь, симптоматическое.