Синдром Кушинга — результат аномально высоких уровней кортизола или др. глюкокортикоидов в крови. М.б. вызван ятрогенными причинами или чрезмерной эндогенной секрецией кортизола вследствие опухоли надпочечников и гиперсекреции кортикотропина (АКТГ) гипофизом (болезнь Кушинга) или опухолью (табл. 1).
а) Этиология. Частая причина синдрома Кушинга — продолжительное введение экзогенных глюкокортикоидных гормонов (особенно в высоких дозах) для лечения лимфопролиферативных заболеваний. Это редко представляет диагностическую проблему, но контроль гипергликемии, гипертензии, набора веса, задержки линейного роста и остеопороза усложняет терапию кортикостероидами.
Эндогенный синдром Кушинга у младенцев часто вызван функционирующей опухолью коры надпочечников. У пациентов с такими опухолями наблюдаются признаки повышенного уровня кортизола наряду с симптомами гиперсекреции др. стероидов (андрогенов, эстрогенов и альдостерона).
Наиболее частая причина эндогенного синдрома Кушинга у детей >7 лет — болезнь Кушинга, при которой избыток АКТГ, секретируемый аденомой гипофиза, вызывает двустороннюю гиперплазию надпочечников (у младенцев встречается редко). Такие аденомы имеют слишком малые размеры (микроаденомы): их сложно обнаружить при помощи визуализирующих методов. Они состоят из хромофобных клеток и часто имеют положительное иммуноокрашивание на АКТГ и его предшественник проопиомеланокортин.
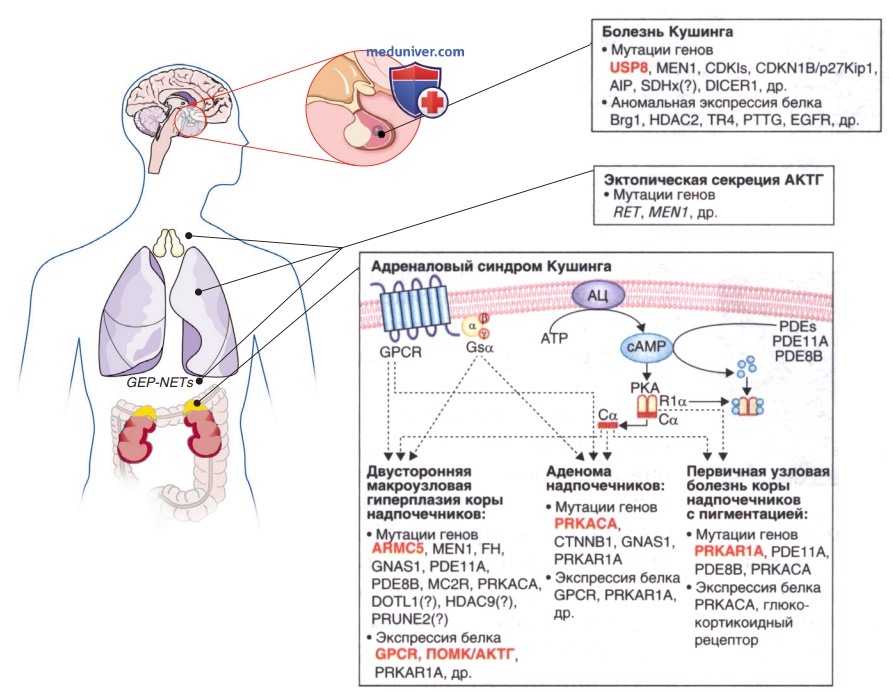
Большинство таких опухолей — спорадические, часть из них возникает в семьях с синдромом семейной изолированной аденомы гипофиза. Этот синдром — результат мутаций гена белка, взаимодействующего с арил-гидрокарбоновым рецептором (встречается при 2% аденом гипофиза). Часто опухоли с мутациями арил-гидрокарбонового рецептора секретируют гормон роста или пролактин, редко — АКТГ. Сходным образом у пациентов с синдромом множественной эндокринной неоплазии 1-го типа (мутации гена менина — MEN1) развиваются опухоли гипофиза (чаще пролактиномы). В процесс могут быть вовлечены и др. гены (рис. 1).
Рисунок 1. Краткое изложение генетических и молекулярных механизмов, вовлеченных в синдром Кушинга. Для каждой причины показаны различные генетические мутации или аномальная экспрессия белка, которые играют определенную роль в патофизиологии. Часто встречающиеся механизмы выделены красным; хорошо описанные механизмы выделены жирным шрифтом, а другие потенциальные механизмы — обычным шрифтом; вопросительный знак указывает на неподтвержденную связь или генетическую предрасположенность. Обратитесь к тексту для объяснения различных генетических дефектов в каждой диагностической категории. АЦ — аденилатциклаза; АКТГ — адренокортикотропный гормон; двусторонняя макроузловая гиперплазия коры надпочечников; Со — каталитическая субъединица протиеинкиназы; GPCR — G-белок-сопряженный рецептор; PDEs — фосфодиэстеразы; РКА — протеинкиназа A; PPNAD — первичная узловая болезнь коры надпочечников с пигментацией; Riα, регуляторная субъединица протеинкиназы lα типа.
АКТГ-зависимый синдром Кушинга также может быть результатом эктопической продукции АКТГ, хотя для детей это нетипично. Эктопическая секреция АКТГ у детей ассоциирована с карциномой островковых клеток ПЖЖ, нейробластомой или ганглионейробластомой, гемангиоперицитомой, опухолью Вильмса и карциноидом вилочковой железы. Гипертензия чаще встречается при синдроме эктопической продукции АКТГ, чем при др. формах синдрома Кушинга. Высокие уровни кортизола могут подавлять активность 11β-гидроксистероид-дегидрогеназы в почках и усиливать свое минералокортикоидное действие (задержка соли).
Некоторые синдромы больше ассоциированы с развитием множественных автономных гиперфункционирующих узелков в ткани коры надпочечников, нежели с одиночными аденомами или карциномами. Во многих случаях они вызваны мутациями генов циклического АМФ-опосредованного сигнального пути, посредством которого АКТГ регулирует секрецию кортизола в норме. Первичная узловая болезнь коры надпочечников с пигментацией — особая форма АКТГ-независимого синдрома Кушинга. Она может возникать как изолированное состояние или как семейное заболевание с др. проявлениями.
Надпочечники имеют небольшой размер и типичные множественные, маленькие (<4 мм в диаметре), пигментированные (черные) узелки, содержащие гигантские клетки с цитоплазмой и липофусцином; между узелками кора атрофирована. Это заболевание надпочечников возникает как компонент комплекса Карни. Комплекс Карни — аутосомно-доминантное заболевание, при котором отмечаются центрофациальные лентиго и синие невусы; сердечные и кожные миксомы; опухоли гипофиза, ЩЖ и яичек; пигментные меланотические шванномы. Комплекс Карни наследуется по аутосомно-доминантному типу, хотя также возникают и спорадические случаи.
Генетические локусы комплекса Карни были картированы в гене регуляторной субъединицы протеинкиназы А 1α типа (PRKAR1A), расположенном на 17q22-24 хромосоме или на 2р16 хромосоме. У пациентов с комплексом Карни и мутациями PRKAR1A первичная узловая болезнь коры надпочечников с пигментацией обычно развивается во взрослом возрасте. У пациентов с заболеванием, картированным на хромосоме 2 (в большинстве спорадических случаев), первичная узловая болезнь коры надпочечников с пигментацией развивается реже и позже.
У детей с изолированной первичной узловой болезнью коры надпочечников с пигментацией редко обнаруживаются мутации в PRKAR1A или развиваются др. проявления комплекса Карни впоследствии. У некоторых пациентов с изолированной первичной узловой болезнью коры надпочечников с пигментацией отмечаются мутации в генах PDE8B или PDE11А, кодирующих различные изоферменты фосфодиэстеразы. Наоборот, при кортизол-секретирующих аденомах были зарегистрированы активирующие соматические мутации в каталитической субъединице протеинкиназы А 1α типа.
В редких случаях при синдроме Мак-Кьюна-Олбрайта возникает АКТГ-независимый синдром Кушинга с узелковой гиперплазией и формированием аденомы (симптомы появляются в младенческом или детском возрасте). Синдром Мак-Кьюна-Олбрайта — результат соматической мутации гена GNAS, кодирующего G-белок — Gsα, посредством которого работает рецептор АКТГ (MCR2). Это приводит к подавлению активности ГТФ и конститутивной активации аденилатциклазы, повышая тем самым уровень циклического АМФ.
При мутации в ткани надпочечников стимуляция секреции кортизола и деления кл. происходит независимо от АКТГ. Др. ткани, в которых могут возникать активирующие мутации: костная ткань (фиброзная дисплазия), половые железы, ЩЖ и гипофиз. Клинические проявления зависят от того, какая ткань была затронута.
Гены, ассоциированные с узелковой гиперплазией коры надпочечников, вызывают гиперактивность сигнального пути АКТГ как за счет конститутивной активации Gsα (синдром Мак-Кьюна-Олбрайта) при уменьшении распада циклического АМФ, что увеличивает его внутриклеточный уровень (мутации PDE8B или PDE11A), так и за счет нарушения регуляции циклического АМФ-зависимого фермента протеинкиназы А (мутации PRKAR1A).
Поражения коры надпочечников, включая диффузную гиперплазию, узелковую гиперплазию, аденому и карциному, могут возникать как часть синдрома множественной эндокринной неоплазии 1-го типа. Это АуД-заболевание, при котором происходит гомозиготная инактивация гена-супрессора опухоли менина (MEN1), расположенного на хромосоме 11q13.
б) Клинические проявления. Признаки синдрома Кушинга выявляются у детей <1 года. У младенцев заболевание имеет более тяжелое течение, а клинические признаки выражены сильнее, чем у более старших детей. Лицо округлое, с выступающими щеками и гиперемией кожи (лунообразное лицо). У детей младшего возраста встречается генерализованное ожирение. У детей с опухолями надпочечников возникают признаки аномальной маскулинизации: гирсутизм лица и тела, оволосение лобка, акне, огрубение голоса и увеличение клитора у девочек.
Отмечается нарушение роста (уменьшение длины тела ниже 3-го процентиля), за исключением случаев, когда значительная вирилизация приводит к нормальному или даже ускоренному росту. Часто возникает гипертензия, которая иногда может приводить к развитию СН. Повышение подверженности инфекциям увеличивает риск сепсиса.
У детей старшего возраста кроме ожирения ведущий признак болезни — низкорослость. Постепенно развивающееся ожирение и замедление или остановка роста могут быть единственными ранними проявлениями синдрома Кушинга. У детей старшего возраста ожирение лица и туловища выражено сильнее по сравнению с конечностями. Типично появление фиолетовых стрий на передней и задней поверхности бедер и животе.
Может наблюдаться задержка полового развития или аменорея у девочек, у которых менархе уже наступило. Отмечается выраженная слабость, головная боль и эмоциональная лабильность, гипертензия и гипергликемия (может прогрессировать до СД). Часто возникает остеопороз, приводящий к патологическим переломам.
в) Лабораторные признаки. В норме уровень кортизола в крови наиболее высокий в 8 ч утра, снижается до <50% к полуночи, за исключением младенцев и детей младшего возраста, у которых суточный ритм не всегда установлен. У пациентов с синдромом Кушинга циркадианный ритм утрачен; уровень кортизола в полночь >4,4 мкг/дл (>120 нмоль/л) свидетельствует в пользу данного диагноза. Получить образцы суточной мочи в амбулаторных условиях достаточно трудно.
Кортизол можно измерить в образце слюны, который легко получить в домашних условиях в определенное время дня. Повышение уровня кортизола в образцах слюны, полученных в ночное время, требует исключения синдрома Кушинга.
Экскреция свободного кортизола с мочой повышена. Она м.б. измерена в образце суточной мочи и выражается как показатель экскретированного кортизола в микрограммах на грамм креатинина. Показатель не зависит от размера тела и полноты сбора мочи.
Используется дексаметазоновый тест с однократным введением препарата. Введение дозы 25-30 мкг/кг (максД 2 мг) в 11 ч вечера приводит к снижению кортизола в плазме до уровня менее 5 мкг/дл (138 нмоль/л) в 8 ч утра следующего дня у ЗЛ, но не у пациентов с синдромом Кушинга. Следует измерять уровень дексаметазона в том же образце крови для обеспечения адекватности дозирования.
Результаты глюкозотолерантного теста обычно выходят за пределы нормы, однако это не имеет диагностического значения. Уровень электролитов сыворотки крови нормальный, но может отмечаться снижение уровня калия (особенно у пациентов с опухолями, которые эктопически секретируют АКТГ).
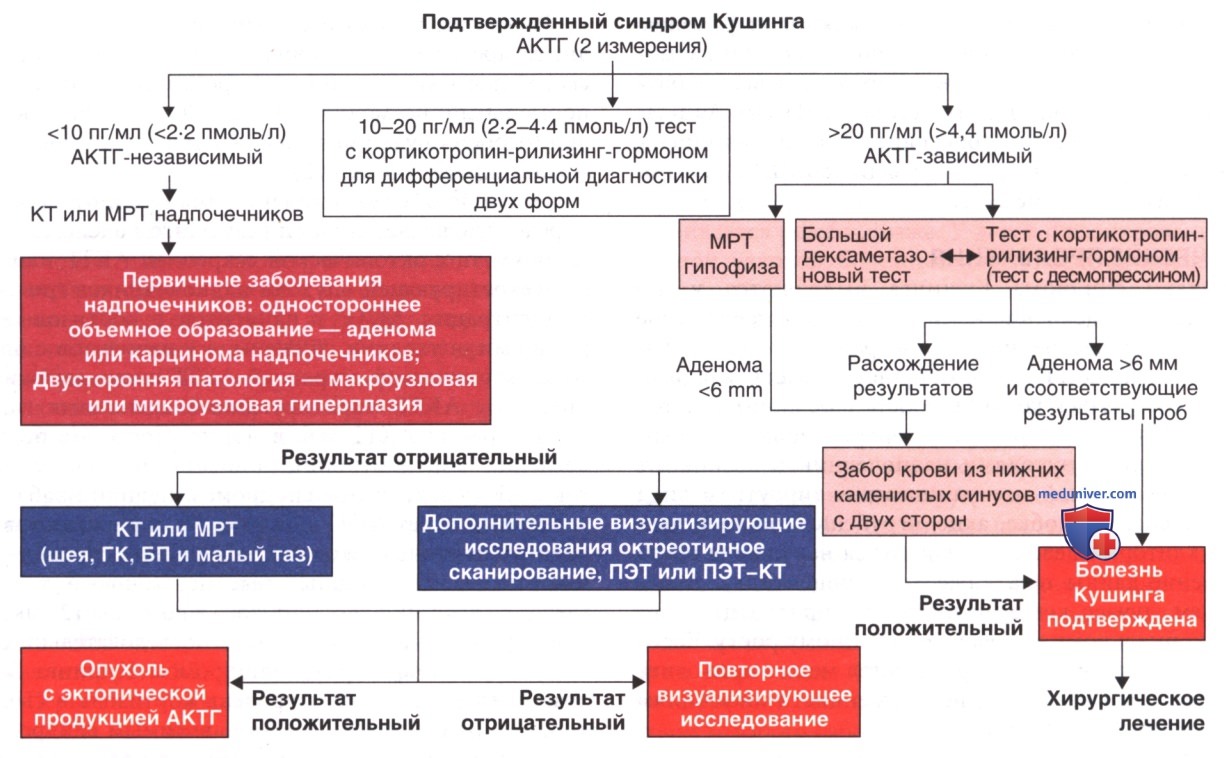
После подтверждения синдрома Кушинга необходимо определить, является ли он результатом аденомы гипофиза, опухоли с эктопической секрецией АКТГ или кортизол-секретирующей опухоли надпочечников (рис. 2). Концентрации АКТГ у пациентов с кортизол-секретирующими опухолями снижены, а у пациентов с опухолями с эктопической секрецией АКТГ очень высоки. У пациентов с АКТГ-секретирующими аденомами гипофиза концентрация АКТГ м.б. в норме. После в/в болюсного введения кортикотропин-рилизинг-гормона у пациентов с АКТГ-зависимым синдромом Кушинга наблюдается усиленный ответ АКТГ и кортизола, а у пациентов с опухолями надпочечников не повышается уровень АКТГ и кортизола.
Рисунок 2. Клиническая схема принятия решений при дифференциальной диагностике подтвержденного синдрома Кушинга различной этиологии. ЗКНКС — забор крови из нижних каменистых синусов с двух сторон; КРГ — кортикотропин-рилизинг-гормон; БДТ — большой дексаметазоновый тест.
Двухэтапный дексаметазоновый тест заключается во введении дексаметазона в дозе 30 и 120 мкг/кг/сут в четыре приема в течение двух последовательных суток. У детей с гипофизарным синдромом Кушинга большая доза препарата снижает уровень кортизола в сыворотке крови. У пациентов с АКТГ-независимым синдромом Кушинга снижение уровня кортизола при введении дексаметазона не наблюдается.
КТ выявляет опухоли надпочечников >1,5 см. С помощью МРТ можно обнаружить АКТГ-секретирующие аденомы гипофиза, но многие из них слишком малы (добавление контраста с гадолинием повышает чувствительность метода). Забор крови из нижних каменистых синусов с двух сторон для измерения концентрации АКТГ до и после введения кортикотропин-рилизинг-гормона требуется для определения расположения опухоли, когда аденома гипофиза не визуализируется. Метод недоступен во многих центрах, а у детей такое обследование низкоспецифично.
Для выявления осложнений болезни Иценко-Кушинга проводится исследование ССС (ЭКГ, по показаниям — ЭхоКГ, холтеровское мониторирование ЭКГ), костно-мышечной (рентгенография грудного и поясничного отделов позвоночника, остеоденситометрия поясничного отдела позвоночника и проксимального отдела бедренной кости) и мочеполовой (УЗИ почек, УЗИ органов малого таза) систем, ЖКТ (ФЭГДС, УЗИ БП)*.
P.S. * КР М3 РФ Болезнь Иценко-Кушинга, 2016
г) Дифференциальная диагностика. Синдром Кушинга можно заподозрить у детей с ожирением (при наличии стрий и гипертензии). У детей с простым ожирением обычно высокий рост, у больных с синдромом Кушинга наблюдается низкорослость или отставание в росте. Уровень экскреции кортизола с мочой при простом ожирении часто повышен, уровни кортизола в образцах слюны, полученных в ночное время, обычно в пределах нормы, а секреция кортизола подавляется при приеме малых доз дексаметазона внутрь.
У пациентов с генерализованной резистентностью к глюкокортикоидам повышаются уровни кортизола и АКТГ без клинических данных синдрома Кушинга. У пациентов может не быть симптомов, иногда выявляется гипертензия, гипокалиемия и преждевременный псевдопубертат. Эти проявления — результат повышения секреции минералокортикоидов и андрогенов надпочечников в ответ на повышение уровней АКТГ. Также выявляются мутации глюкортикоидного рецептора.
д) Лечение. Микрохирургическая операция на гипофизе с трансфеноидальным доступом — метод выбора при гипофизарной болезни Кушинга у детей. Общий показатель эффективности на протяжении периода последующего наблюдения <10 лет составляет 60-80%. Низкие концентрации кортизола в сыворотке крови и моче в послеоперационном периоде — предикторы длительной ремиссии. Для лечения рецидивов проводят повторную операцию или облучение гипофиза.
Для лечения болезни Кушинга у взрослых применялся ципрогептадин — антагонист рецепторов серотонина центрального действия, который блокирует высвобождение АКТГ; после прекращения лечения ремиссия нестойкая. Этот препарат у детей применяется редко. В предоперационном периоде для нормализации уровней кортизола в крови и снижения числа осложнений и смертности применяются ингибиторы стероидогенеза в надпочечниках (метирапон, кетоконазол, аминоглютетимид, этомидат, митотан). Иногда применяется антагонист глюкокортикоидных рецепторов мифепристон.
Аналог соматостатина пасиреотид может подавлять секрецию АКТГ. Он одобрен для применения у взрослых с заболеванием, которое сохраняется после операции, или для пациентов с противопоказаниями к хирургическому лечению. Каберголин (>16 лет) рекомендован при неэффективности нейрохирургического лечения в монотерапии и в комбинациях с другими препаратами вне зависимости от исходного уровня пролактина*.
P.S. * КР МЗ РФ Болезнь Иценко-Кушинга, 2016.
Если аденома гипофиза не отвечает на лечение или если АКТГ секретируется эктопической метастатической опухолью, может потребоваться удаление надпочечников. Процедура проводится лапароскопически. Адреналэктомия приводит к увеличению секреции АКТГ нерезецированной аденомой гипофиза, о чем свидетельствует выраженная гиперпигментация (синдром Нельсона).
Лучевая терапия (радиотерапия, радиохирургия) рекомендована пациентам при неэффективности или невозможности нейрохирургического лечения.
Ведение пациентов, перенесших адреналэктомию, требует адекватной заместительной терапии кортикостероидами в предоперационном и послеоперационном периоде. Наличие опухолей, продуцирующих кортикстероиды, приводит к атрофии нормальной ткани надпочечников: для восстановления гипоталамо-гипофизарно-надпочечниковой оси необходима заместительная терапия кортизолом (10 мг/м2/сут в три раздельных приема сразу после операции). Послеоперационные осложнения: сепсис, панкреатит, тромбоз, плохое заживление ран и внезапные обмороки, особенно у младенцев с синдромом Кушинга.
Наблюдается усиленный догоняющий рост, прогрессирование полового развития и увеличение плотности костей, однако костная плотность остается аномальной, часто имеется риск низкорослости во взрослом возрасте. Ведение пациентов с опухолями коры надпочечников рассматривается в отдельной статье на сайте - просим Вас пользоваться формой поиска по сайту выше.