Краниосиностоз определяется как преждевременное закрытие черепных швов и делится на первичный/вторичный. Характеризуется разл. типами аномалий формы черепа. Первичный краниосиностоз возникает при закрытии одного/нескольких швов вследствие аномалий развития черепа, тогда как вторичный краниосиностоз — из-за недостаточности роста и развития ГМ. Заболеваемость первичным краниосиностозом 1:2000 новорожденных.
У большинства детей причина неизвестна, однако генетические синдромы составляют 10-20% случаев. Во многих случаях важную роль играют деформации при затылочной и лобной плагиоцефалии. Раннее выявление задней формы имеет решающее значение и позволяет провести эффективное физиотерапевтическое лечение кривошеи и др. позиционных асимметрий, приводящих к плагиоцефалии.
а) Развитие и этиология. Кости черепа хорошо развиты к 5-му месяцу беременности (лобные, теменные, височные и затылочные) и разделены швами и родничками. ГМ быстро растет в первые несколько лет жизни и обычно не испытывает затруднений из-за эквивалентного роста по линиям шва. Причина краниосиностоза неизвестна, но основной гипотезой является аномальное развитие основания черепа, которое создает давление на ТМО, приводящее к нарушению нормального развития черепных швов. Генетические факторы выявлены при некоторых изолированных и при многих синдромных краниосиностозах (табл. 7 и рис. ниже). Некорректированный гипертиреоз у матери может привести к развитию данной патологии.
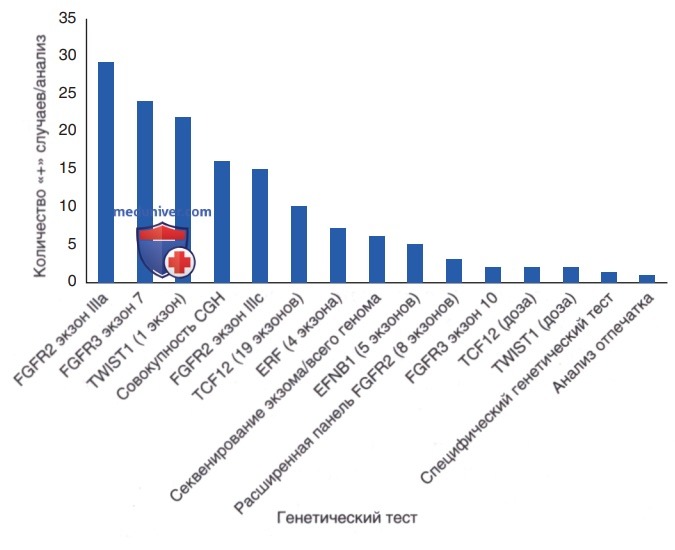
Генетическое тестирование при краниосиностозе. Тесты расположены иерархически: слева те, что дают наибольшее число диагнозов.
б) Клинические проявления и лечение. Большинство случаев краниосиностоза проявляются при рождении и характеризуются деформацией черепа, которая является прямым результатом преждевременного сращения швов. Пальпация шва выявляет выступающий костный гребень, сращение шва м.б. подтверждено простыми рентгенограммами черепа, КТ и сканированием костей (табл. 8).
Преждевременное закрытие сагиттального шва приводит к образованию длинного и узкого черепа — скафоцефалии, наиболее распространенной формы краниосиностоза. Скафоцефалия характеризуется выступающим затылком, широким лбом и неболыпим/отсутствующим передним родничком. Это заболевание носит спорадический характер, чаще встречается у мальчиков и вызывает трудности во время родов из-за головно-тазовой диспропорции. Скафоцефалия не ведет к повышению ВЧД/гидроцефалии, результаты неврологического обследования пациентов в пределах нормы.
Лобная плагиоцефалия является следующей наиболее распространенной формой краниосиностоза и характеризуется односторонним уплощением лба, подъемом ипсилатеральной орбиты и брови, выступающим ухом на соответствующей стороне. Это заболевание чаще встречается у девочек и является результатом преждевременного сращения коронарного и сфенофронтального швов. Хирургическое вмешательство дает хороший косметический результат. При отсутствии закрытых швов при визуализации первостепенное значение имеют признаки позиционной деформации.
Затылочная плагиоцефалия чаще всего является результатом позиционирования в младенчестве и встречается у неподвижного ребенка/ребенка с инвалидностью, но слияние/склероз лямбдоидного шва может вызвать одностороннее затылочное уплощение и выпячивание ипсилатеральной лобной кости.
Тригоноцефалия — редкая форма краниосиностоза, вызванная преждевременным сращением метопического шва. Эти дети имеют килевидный лоб и гипотелоризм, подвержены риску развития сопутствующих аномалий переднего мозга. Более мягкие формы раннего сращения метопического гребня встречаются чаще.
Туррицефалия (оксицефалия) — конусообразная деформация головы из-за преждевременного сращения коронарных, а часто и сфенофронтальных и фронтоэтмоидальных швов. При деформации черепа по типу трилистника он напоминает лист клевера. Больные дети имеют значительно выступающие височные кости с сужением остальной части черепа. Гидроцефалия является распространенным осложнением в данном случае.
Преждевременное сращение только одного шва редко вызывает неврологический дефицит. В этой ситуации единственным показанием к операции являются косметические проблемы, а прогноз зависит от измененного шва и степени обезображивания. Неврологические осложнения, включая гидроцефалию и повышение ВЧД, чаще возникают при преждевременном сращении >2 швов, в этом случае оперативное вмешательство необходимо. Роль усилий по ранней репозиции и терапии кривошеи, а также использование краниальных приспособлений выходят за рамки настоящего обзора.
Наиболее распространенными генетическими нарушениями, сопровождающимися краниосиностозом, являются синдромы Крузона, Аперта, Карпентера, Хотцена и Пфайффера. Синдром Крузона характеризуется преждевременным краниосиностозом и наследуется как АуД-признак. Форма головы зависит от времени и порядка сращения швов, но чаще всего уменьшен переднезадний диаметр/брахицефалия, возникающая в результате двустороннего закрытия коронарных швов. Орбиты недоразвиты, выявляется глазной проптоз. Типичными являются гипоплазия верхней челюсти и орбитальный гипертелоризм.
Синдром Аперта имеет много общих черт с синдромом Крузона. Синдром Аперта обычно возникает спорадически, хотя может иметь место АуД-наследование. Это связано с преждевременным закрытием нескольких швов, включая корональные, сагиттальные, плоскоклеточные и лямбдоидные швы. Лицо, как правило, асимметричное, а глаза менее проптотичны, чем при синдроме Крузона. Синдром Аперта характеризуется синдактилией 2, 3 и 4-го пальцев, которые м.б. соединены с большим и 5-м пальцами. Подобные аномалии часто встречаются на ногах. У всех пациентов наблюдается прогрессирующее обызвествление и сращение костей кистей, стоп и ШОП.
Синдром Карпентера наследуется как АуР-состояние, и многочисленные сращения швов, как правило, приводят к деформации черепа по типу трилистника. Синдактилия мягких тканей кистей и стоп наблюдается у всех, а умственная отсталость является распространенным явлением. Дополнительные, но менее распространенные аномалии включают ВПС, помутнения роговицы, диспластический коксартроз и деформацию коленного сустава.
Синдром Хотцена характеризуется асимметричным краниосиностозом и плагиоцефалией. Это заболевание является наиболее распространенным из генетических синдромов и наследуется как АуД-признак. Характерные симптомы: асимметрия лица, птоз век, укорочение пальцев и синдактилия мягких тканей 2-го и 3-го пальцев.
Синдром Пфайффера чаще всего сопровождается туррицефалией. Глаза выпуклые и широко расставленные, а большие пальцы рук и ног короткие и широкие. Встречается частичная синдактилия мягких тканей. Большинство случаев, по-видимому, носят спорадический характер, но сообщалось об АуД-наследовании. Показано, что мутации семейства генов рецептора фактора роста фибробластов вызывают фенотипически специфичные типы краниосиностоза.
Мутации гена FGFR1, расположенного на хромосоме 8, приводят к развитию синдрому Пфайффера; аналогичная мутация гена FGFR2 вызывает синдром Аперта. Идентичные мутации гена FGFR2 могут приводить как к фенотипам синдрома Пфайффера, так и к фенотипам синдрома Крузона.
Каждый из генетических синдромов создает риск дополнительных аномалий, включая гидроцефалию, повышение ВЧД, папиллезию, атрофию зрительного нерва, возникающую в результате аномалий зрительного отверстия, респираторные проблемы вследствие искривления носовой перегородки/атрезии хоан, а также нарушения речи и глухоту. Краниоэктомия является обязательной для лечения повышенного ВЧД, а работа мультидисциплинарной бригады специалистов черепно-лицевой области имеет значение при долгосрочном наблюдении за пострадавшими детьми. Краниосиностоз может иметь благоприятный исход при хирургическом лечении, приводящем к снижению смертности, особенно у несиндромных детей.
Видео швы и роднички головки плода - плод как объект родов