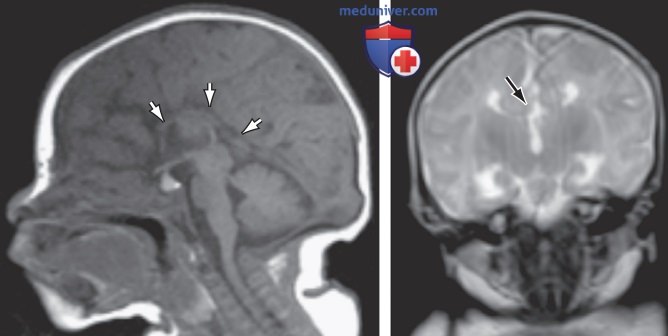
К агенезии мозолистого тела относится гетерогенная группа нарушений, которые по выраженности варьируют от тяжелых интеллектуальных, неврологических нарушений до бессимптомных и пациентов с нормальным интеллектом (рис. ниже).
Агенезия мозолистого тела показана на магнитно-резонансной томограмме головного мозга. Сагиттальный (слева) и корональный (справа) снимки младенца демонстрируют полное отсутствие структуры белого в-ва центральной сагиттальной области (слева, стрелки). Корональный вид (справа) демонстрирует (несмотря на некоторый артефакт движения) отсутствие структуры, соединяющей два полушария (область под стрелкой)
Мозолистое тело развивается из спаечной пластинки, которая находится в непосредственной близости от передней нейропоры. Агенезию мозолистого тела может вызвать прямое повреждение спаечной пластинки, нарушение генетической сигнализации, определяющей и организующей эту область в раннем эмбриогенезе. При изолированной агенезии мозолистого тела в остальном пациент м.б. здоров.
Когда процесс сопровождается ВПР ГМ вследствие дефектов миграции клеток, таких как гетеротопии, полимикрогирия и пахигирия (широкие, глубокие извилины), пациенты часто имеют значительные неврологические отклонения, включая умственную отсталость, микроцефалию, гемипарез/диплегию и судороги.
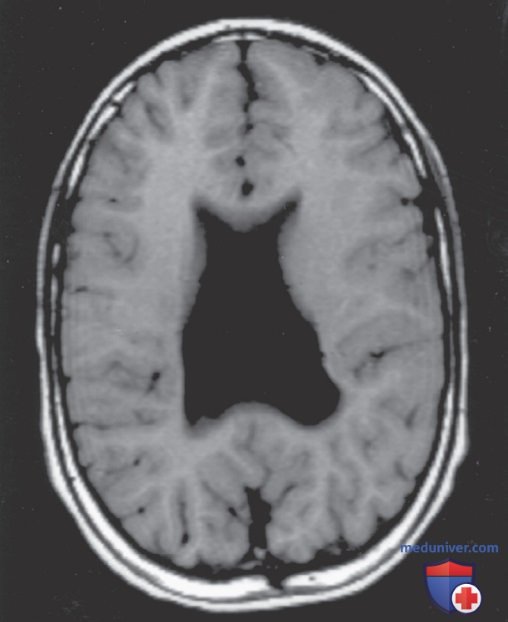
Анатомические особенности агенезии мозолистого тела лучше всего визуализируются на МРТ и представлены широко разделенными лобными рогами с аномально высоким положением третьего желудочка между боковыми желудочками. МРТ определяет степень дефекта мозолистого тела. Его отсутствие м.б. унаследовано как Х-сцепленный рецессивный признак/АуД-признак, реже как АуР-признак.
Это состояние м.б. обусловлено специфическими хромосомными нарушениями, в частности трисомией 8 и трисомией 18. Мутации одного гена описаны в нескольких генах, вызывающих агенезию мозолистого тела. Выявлены вариации числа копий (делеции), но обычно при наличии др. сопутствующих аномалий. Агенезия мозолистого тела наблюдается при некоторых метаболических нарушениях (табл. 3).
Синдром Айкарди представляет собой сложное расстройство, которое затрагивает многие системы и обычно сопровождается агенезией мозолистого тела, характерными хориоретинальными лакунами и инфантильными спазмами. Пациентки почти все женского пола, что свидетельствует о генетической аномалии Х-хромосомы (она м.б. смертельной для мужского пола во в/утробный период).
Приступы проявляются в течение первых нескольких месяцев и, как правило, устойчивы к противосудорожным ЛП. ЭЭГ регистрирует независимую активность от обоих полушарий в результате отсутствия мозолистого тела и часто гемигипсаритмию. Все пациенты имеют тяжелую умственную отсталость, возможно наличие аномальных позвонков — сросшиеся/частично развитые (гемивертебры). Аномалии сетчатки, включая ограниченные ямки/лакуны и колобому диска зрительного нерва, являются наиболее характерными признаками синдрома Айкарда.
Кольпоцефалия относится к патологическому увеличению затылочных рогов желудочковой системы и м.б. выявлена уже в эмбриональном периоде. Ей может сопутствовать агенезия мозолистого тела, но может возникать изолированно. Кальпоцефалия характерна для микроцефалии. Данный синдром можно наблюдать при анатомической мегалэнцефалии, напр. при синдроме Сотоса.
а) Голопрозэнцефалия. Голопрозэнцефалия — дефект развития ГМ, возникающий в результате аномального формирования просенсефалона и неадекватной индукции структур переднего мозга. Аномалия классифицируется на три группы: алобарную, полулобарную и долевую, в зависимости от степени нарушения расщепления (рис. ниже). Четвертый тип, средний вариант межполушарного слияния/синтелэнцефалия, включает сегментарную область нерасщепления, фактически несепарацию, задней лобной и теменной долей.
Лицевые аномалии, включая циклопию, синофтальмию, цебоцефалию, единственную ноздрю, хоанальную атрезию, одиночный центральный резец и премаксиллярную агенезию, часто встречаются в тяжелых случаях, т.к. прехордальная мезодерма, которая индуцирует вентральный прозэнцефалон, ответственна за индукцию срединных лицевых структур. Легкие аномалии лица наблюдаются при нетяжелых клинических формах. Алобарная голопрозэнцефалия новорожденных характеризуется наличием единственного желудочка, отсутствием серпа и неразделенных глубоких мозговых ядер.
Необходимо соблюдать осторожность при постановке диагноза, основанного только на желудочковых аномалиях. Наличие неразделенных срединных глубинных структур ГМ, таких как хвост, путамен, бледный шар и гипоталамус, является основанием для диагноза.
Дети с алобарным типом имеют высокий уровень смертности, но некоторые живут годами. Смертность и заболеваемость с более легкими типами изменчивы, а течение менее тяжелое. Во всех случаях необходимо соблюдать осторожность при прогнозировании тяжелых исходов. Частота голопрозэнцефалии 1:5000-16000 новорожденных. Пренатальный диагноз м.б. подтвержден УЗИ после 10-й недели беременности при тяжелых типах, но МРТ плода в более позднем гестационном возрасте дает гораздо большую анатомическую, а следовательно, диагностическую точность.
Причина голопрозэнцефалии часто неясна. По-видимому, существует связь с материнским СД. Хромосомные аномалии, включая делеции хромосом 7q и 3р, 21q, 2р, 18р и 13q, а также трисомии 13 и 18, составляют >50% всех случаев. Мутации в гене sonic Hedgehog на 7q также м.б. причиной этой патологии. Исследование генов показало 14 причин, обусловленных одним геном. Клинически важно искать сопутствующие аномалии, т.к. голопрозэнцефалия характерна для многих синдромов.