Под гипопитуитаризмом понимают недостаток одного или нескольких гормонов гипофиза. У детей с этим заболеванием наблюдается задержка роста в постнатальном периоде и недостаточность др. гормонов, что приводит к необходимости заместительной гормональной терапии. Предполагается, что частота врожденного гипопитуитаризма составляет от 1:4000 до 1:10 000 живорожденных. Существует эпидемиологическая ассоциация между развитием гипопитуитаризма и родами в тазовом предлежании, однако причинно-следственные связи не изучены.
С расширением знаний о генах, которые влияют на развитие гипофиза или выработку гормонов, все большую часть случаев можно отнести к конкретным генетическим нарушениям. Мутации в семи генах-кандидатах составляют 13% случаев изолированного дефицита гормона роста (ИДГР) и 20% случаев множественного дефицита гормонов гипофиза (МДГГ). Вероятность обнаружения мутаций увеличивается у ребенка от близкородственного брака, а также при возникновении заболевания у братьев и сестер или в ряде поколений; однако в большинстве случаев ИДГР и МДГГ идентифицировать конкретную генетическую причину не удается.
Гены, гормональные фенотипы, ассоциированные с ними заболевания, а также типы передачи доказанных генетических нарушений приведены в табл. 1 и табл. 3-5. Приобретенный гипопитуитаризм обычно развивается позже и имеет различные причины (табл. 2).
а) Множественный дефицит гормонов гипофиза:
1. Генетические формы (см. табл. 1). Последовательная экспрессия транскрипционных активирующих факторов определяет дифференцировку и пролиферацию клеток передней доли гипофиза. Эти белки являются членами большого семейства ДНК-связывающих белков, напоминающих гомеобокс-содержащие гены. Вследствие мутаций возникают разные формы МДГГ. Гены PROP1 и POU1F1 экспрессируются при развитии гипофиза довольно поздно и только в кл. передней доли, мутации этих генов приводят к развитию гипопитуитаризма без аномалий развития других органов и систем. Гены HESX1, LHX3, LHX4, ОТХ2, SOX3 и PITX2 экспрессируются на более ранних стадиях и отвечают не только за развитие гипофиза.
Мутации в этих генах приводят к развитию фенотипа, выходящего за рамки гипопитуитаризма и включающего аномалии развития др. органов при различной степени выраженности гипопитуитаризма.
- PROP1. PROP1 (предшественник PIT1) найден в ядрах клеток соматотрофов, лактотрофов и тиреотрофов. Его роль заключается в запуске экспрессии гена POU1F1 и снижении экспрессии гена HESX1. Несмотря на то, что у большинства пациентов с МДГГ мутации в генах не выявляются, именно мутации PROP1 являются наиболее частой причиной рецессивного МДГГ и встречаются в 10 раз чаще, чем мутации других генетических факторов транскрипции гипофиза, вместе взятых. Чаще всего встречаются делеции одной или двух пар оснований во 2-м экзоне, реже — миссенс-, нонсенс-мутации и мутации сайта сплайсинга. Дефицит гормонов передней доли гипофиза редко проявляется в неонатальном периоде.
Средний возраст диагностики дефицита ГР составляет ~6 лет. По сравнению с дефицитом ГР выявление недостаточности ТТГ, ЛГ, ФСГ и АКТГ происходит позже. У большинства пациентов размер передней доли гипофиза уменьшен, но у некоторых наблюдается его прогрессирующее увеличение.
- POU1F1 (PIT1). POU1F1 (ранее PIT1) идентифицирован как ядерный белок, который связывается с промоторами ГР и пролактина. Это необходимо для развития и созревания функционирования соматотрофов, лактотрофов и тиреотрофов. Доминантные и рецессивные мутации гена POU1F ответственны за тотальный дефицит гормона роста, пролактина и вариабельный дефицит ТТГ. У пациентов с этой мутацией наблюдается нормальное развитие плода, однако возникает резкая задержка роста на первом году жизни. Благодаря нормальной выработке ЛГ и ФСГ половое развитие начинается самостоятельно, хотя и несколько позже обычного. Пациенты не подвержены риску развития недостаточности АКТЕ Размер передней доли гипофиза варьирует от нормального до уменьшенного.
- HESX1. Ген HESX1 экспрессируется в предшественниках всех 5 типов кл. передней доли гипофиза на ранних этапах эмбрионального развития. Мутации в этом гене приводят к гетерогенным фенотипам с дефектами развития зрительного нерва и гипофиза. Может наблюдаться аплазия или гипоплазия передней доли гипофиза, а задняя доля может находиться в правильном положении или быть эктопированной. Пациенты могут иметь ИДГР или МДГГ с синдромом гипоплазии зрительного нерва или без него. Сочетание недоразвития прозрачной перегородки с гипоплазией зрительного нерва и гипофизарной недостаточностью также называют септооптической дисплазией. Однако у подавляющего большинства пациентов с синдромом гипоплазии зрительного нерва мутаций гена HESX1 не обнаруживают.
- LHX3 и LHX4. Фенотип при рецессивных инактивирующих мутациях гена LHX3 напоминает фенотип при мутациях гена PROP1. Возникает недостаточность ГР, пролактина, ТТГ, ЛГ и ФСГ, но не АКТЕ У некоторых наблюдается увеличение аденогипофиза. Дополнительные необычные признаки, описанные у таких пациентов, — короткая шея и ригидность мышц шейного отдела позвоночника. Доминантно наследуемые мутации в структурно схожем гене LHX4 всегда вызывают дефицит ГР с различной выраженностью недостаточности ТТГ и АКТЕ Дополнительные находки могут включать: очень маленькую V-образную ямку гипофиза, мальформацию Киари I, эктопию задней доли гипофиза.
2. Другие врожденные формы. Гипоплазия гипофиза может возникать изолированно или в сочетании с более тяжелыми аномалиями развития, такими как анэнцефалия или голопрозэнцефалия. Аномалии средней линии лица (расщелина губы, нёба) или наличие единственного центрального резца указывают на высокую вероятность дефицита ГР или других гормонов передней или задней доли гипофиза (рис. 1). Не менее 12 генов вовлечено в сложную генетическую этиологию голопроэнцефалии. Синдром Паллистера-Холла обусловлен доминантными инактивирующими мутациями гена GLI3. Аплазия гипофиза сочетается с гипоталамической гамартомой, полидактилией, дисплазией ногтей, расщелиной надгортанника, атрезией ануса и аномалиями сердца, легких, почек. Сочетание анофтальмии и гипопитуитаризма связано с мутациями генов SIX6, SOX2, и ОТХ2.
Рисунок 1. Единственный срединный верхнечелюстной центральный резец в возрасте 16 мес.
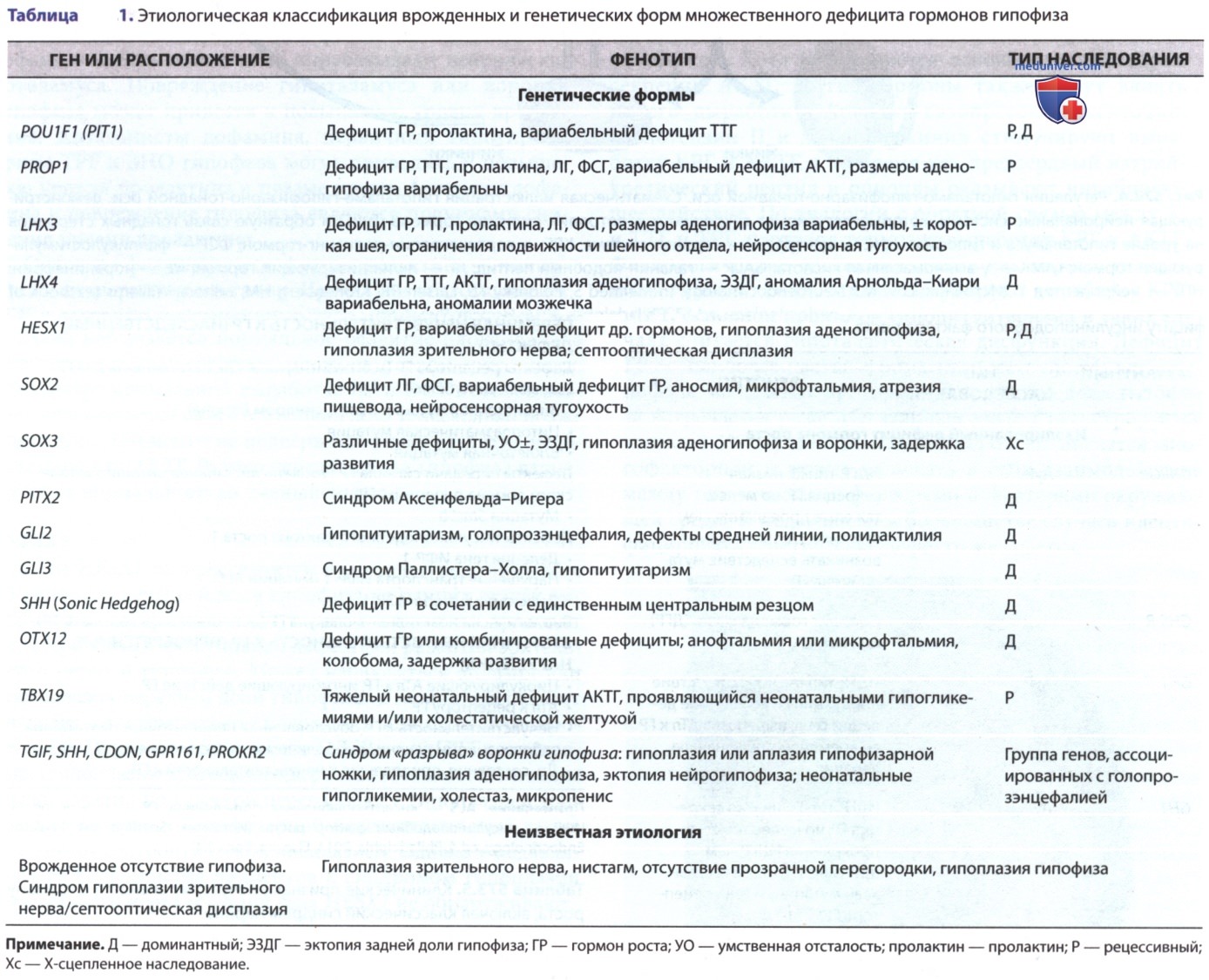
Синдром гипоплазии зрительного нерва, или септооптическая дисплазия, может быть обнаружен при обследовании младенца по поводу нистагма или нарушений зрения. Нейровизуализация выявляет аномалии зрительного нерва и головного мозга, что может быть ассоциировано с дефицитом гормонов передней и/или задней доли гипофиза в 75% случаев (рис. 2). Несмотря на наличие у этих пациентов триады (гипоплазия аденогипофиза и воронки гипофиза, эктопия нейрогипофиза) по данным МРТ, основной причиной гипопитуитаризма в таких случаях считается гипоталамическая дисфункция. Дефицит ГР — наиболее частая форма недостаточности аденогипофиза, выпадение др. гормонов передней доли гипофиза встречается реже. Несахарный диабет регистрируется только в 5% случаев.
Рисунок 2. Септооптическая дисплазия с агенезией прозрачной перегородки. На сагиттальном Т1-взвешенном магнитно-резонансном изображении показано опущение свода (стрелка). Гипоплазия зрительного нерва (короткая стрелка); нейрогипофиз не визуализируется.
Этиология, вероятно, является многофакторной и может включать в себя взаимодействие между генетическими факторами и факторами окружающей среды. В подавляющем большинстве случаев идентифицировать генетические дефекты не удается.
Тяжелый МДГГ с ранним началом включает в себя дефицит АКТГ и часто ассоциирован с триадой (гипоплазия аденогипофиза, аплазия или гипоплазия воронки гипофиза, эктопия нейрогипофиза) по данным МРТ. Большинство случаев носят спорадический характер; заболевание преобладает у мужского пола. Некоторые случаи ассоциированы с аномалиями гена SOX3, расположенного на X-хромосоме.
3. Приобретенные формы. Дефицит гипофизарных гормонов может быть следствием любых повреждений гипоталамуса, воронки гипофиза или его передней доли (см. табл. 2). Поскольку такие поражения не избирательны, обычно возникает множественный дефицит гормонов. При приобретенном гипопитуитаризме несахарный диабет встречается чаще, чем при врожденном. Наиболее частой причиной повреждения является краниофарингиома. Причинами разрушения гипоталамо-гипофизарной структуры могут также быть герминома, эозинофильная гранулема (гистиоцитоз), туберкулез, саркоидоз, токсоплазмоз, менингит, абсцесс гипофиза и аневризмы.
Дети, получавшие лучевую терапию по поводу опухолей ЦНС или носоглотки, подвергаются повышенному риску развития дефицита ГР и др. тропных гормонов в случае, если область облучения включает гипоталамус и/или гипофиз, даже если сама опухоль гипоталамо-гипофизарной системы удалена. Степень риска и время возникновения дефицита гормонов гипофиза зависит от дозы облучения гипоталамо-гипофизарной области и длительности периода после завершения лучевой терапии. Высокие дозы облучения (>50 Гр) могут вызвать развитие дефицита ГР в течение 1 года после облучения, тогда как дефицит др. гормонов аденогипофиза может проявиться позже.
Продукция ГР, по всей видимости, особенно уязвима к воздействию облучения даже в низких дозах, в то время как дефициты АКТГ, гонадотропинов и ТРГ (тиреотропин-рилизинг-гормон)/ТТГ возникают с уменьшающейся частотой и, как правило, при более высоких дозах облучения. Воздействие только облучения обычно не приводит к развитию несахарного диабета. ЧМТ, включая травмы вследствие жестокого обращения, автомобильных аварий и хронические повторяющиеся травмы головы, все чаще признаются причиной нарушения функции гипофиза вследствие повреждения гипофиза, его воронки или гипоталамуса.
б) Изолированный дефицит гормона роста и нечувствительность к гормону роста:
1. Генетические формы дефицита гормона роста. ИДГР может быть вызван аномалиями рецептора ГР-рилизинг-гормона, гена ГР и генов, расположенных на Х-хромосоме (см. табл. 3).
- Рецептор ГР-рилизинг-гормона. Рецессивные инактивирующие мутации генов рецептора ГР-рилизинг-гормона препятствуют пролиферации соматотрофов во время развития гипофиза и нарушают регуляцию наиболее важных сигналов для высвобождения ГР. Передняя доля гипофиза уменьшена в размерах, что соответствует представлениям о том, что соматотрофы занимают >50% объема гипофиза. Имеется умеренная задержка роста плода и резкая задержка роста в постнатальном периоде.
- GH1. Ген GH1 — один из пяти генов кластера, расположенного на хромосоме 17q22-24. Этот кластер возникает в результате последовательных дупликаций предкового гена ГР. Неравный кроссинговер при мейозе ведет к различным делециям в генном кластере. Малые делеции (<10 kb) приводят к выпадению только гена GH1, тогда как большие делеции (45 kb) захватывают один или нескольких соседних генов (CSL, CS1, GH2, и CS2). Делеция только GH1 или GH1 в сочетании с одним или несколькими соседними генами одинаково сказывается на росте. Утрата генов CS1, GH2 и CS2 без потери GH1 вызывает дефицит хорионического соматомаммотропина и плацентарного ГР в материнском кровотоке, но не приводит к задержке внутриутробного или постнатального роста.
Большинство детей с делециями гена GH1 очень хорошо отвечают на терапию рекомбинантным ГР, но у некоторых образуются АТл к ГР, и они перестают расти.
- Сцепленный с Х-хромосомой изолированный дефицит гормона роста. Два локуса на Х-хромосоме ассоциированы с гипопитуитаризмом. Первый находится на Xq21.3-q22 в регионе гена тимидинкиназы Брутона. Мутации в этой области приводят к гипогаммаглобулинемии и ИДГР. Второй локус картирован на Xq24-q27.1, в области, содержащей ген фактора транскрипции SOX2. Аномалии в этом месте связаны с ИДГР в сочетании с умственной отсталостью, а также с МДГГ и триадой симптомов (гипоплазия гипофиза, аплазия воронки и эктопия задней доли гипофиза) при МРТ.
2. Приобретенные формы. Гипоталамическая регуляция секреции ГР более чувствительна к внешним воздействиям, чем другие гипоталамо-гипофизарные системы. Признанными причинами приобретенного дефицита ГР являются лучевая терапия ЗНО, менингит, гистиоцитоз и травма.
Дети, получающие лучевую терапию при опухолях ЦНС и лейкемии, а также облучение всего тела перед трансплантацией гемопоэтических стволовых клеток, подвержены риску развития дефицита ГР. Облучение позвоночника приводит к непропорциональному замедлению роста туловища; этот дефект невозможно исправить лечением гормоном роста. Во время облучения или химиотерапии (ХМТ) рост обычно замедляется, может улучшиться в течение 1-2 лет после завершения лечения, после чего скорость роста вновь снижается в связи с развитием дефицита ГР. Доза и продолжительность лучевой терапии — решающие факторы риска гипопитуитризма. После 5 лет терапии в общей дозе >35 Гр дефицит ГР развивается практически всегда. Дозы 20 Гр оказывают меньшее повреждающее действие.
Чаще всего возникает дефицит ГР, но возможна недостаточность также ТТГ и АКТГ. Облучение области головы может привести к преждевременному половому развитию в сочетании с дефицитом ГР. В таких случаях у детей в возрасте 8-10 лет с признаками полового развития скорость роста может соответствовать хронологическому возрасту, но быть ниже пубертатной.
в) Нечувствительность к гормону роста:
1. Нарушения рецептора гормона роста. Нечувствительность к ГР вызвана нарушением механизмов действия ГР, следующих за его продукцией (табл. 4). Синдром Ларона связан с мутациями в гене рецептора ГР. Дети с этим заболеванием клинически напоминают пациентов с тяжелым ИДГР. Длина тела при рождении обычно на 1 SD ниже средней, а низкорослость, притом тяжелой степени, наблюдается уже на 1-м году жизни. Базальные и стимулированные уровни ГР обычно высокие, а уровни ИФР-1 — низкие. Рецептор ГР имеет внеклеточный ГР-связывающий домен, трансмембранный и внутриклеточный сигнальный домены. Большинство мутаций в гене рецептора ГР приводят к нарушению способности внеклеточного домена связывать ГР.
Активность сывороточного ГР-связывающего белка, представляющего собой циркулирующую форму мембранного рецептора ГР, обычно низкая. Более редкие мутации могут нарушить фиксацию рецептора, лишенного трансмембранного домена, на плазматической мембране. В этих случаях активность циркулирующего ГР-связывающего белка нормальная или высокая. Мутации внутриклеточного домена приводят к нарушению JAK/STAT передачи сигнала.
2. Типы пострецепторной нечувствительности к гормону роста. В некоторых случаях тяжелая низкорослость у детей с высокими уровнями ГР и низкими — ИФР-1, а также нормальными уровнями ГР-связывающещего белка обусловлена нарушением процессов, следующих после связывания ГР и активации рецептора ГР. Так, мутации в гене, кодирующем трансдуктор сигнала и активатор транскрипции 5b (STAT5b), вызывают нарушение роста, схожее с синдромом Ларона. Помимо низкорослости наблюдаются иммуннодефицит и хронические легочные инфекции, так как STAT5b играет важную роль в передаче сигнала цитокинами-интерлейкинами.
- Нарушения гена инсулиноподобного фактора роста 1. Дефекты гена ИФР-1 приводят к резкой задержке роста плода и постнатальной низкорослости. У пациентов с делецией экзона и миссенс-мутацией в гене наблюдается также микроцефалия, умственная отсталость и глухота. Ожидается, что такие пациенты ответят на лечение рекомбинантным ИФР-1.
- Нарушения белка, связывающего инсулиноподобный фактор роста. Мутация гена, кодирующего кислотно-лабильную субъединицу циркулирующего комплекса с молекулярной массой 165 кДа, состоящего из ИФР-1, ИФР-СБЗ, кислотно-лабильной субъединицы, ассоциирована с низкорослостью. У пациентов с такой мутацией в гомозиготном состоянии общий уровень ИФР-1 очень низкий, а лечение ГР не приводит к его повышению и увеличению скорости роста.
- Нарушения гена рецептора инсулиноподобного фактора роста 1. Мутации в гене рецептора ИФР-1 также приводят к снижению пренатального и постнатального роста. Клинические проявления мягче, чем при отсутствии ИФР-1: конечный рост пациентов может быть близок к среднему, нет умственной отсталости или глухоты.
г) Клинические проявления:
1. Врожденный гипопитуитаризм. Длина и МТ при рождении у ребенка с гипопитуитаризмом обычно нормальные, хотя у пациентов с МДГГ и дефектами генов GH1 или GHR длина тела новорожденного в среднем на 1 SD ниже средней. При тяжелых нарушениях продукции или действия ГР рост ребенка к концу первого года жизни более чем на 4 SD ниже среднего. Менее выраженная недостаточность ГР приводит лишь к замедлению темпов роста ниже 25-го процентиля для возраста, а развитие низкорослости происходит постепенно. Задержка закрытия зон роста позволяет пациентам расти дольше обычного. Признаки, характерные для нечувствительности к ГР, включая синдром Ларона, представлены в табл. 5.
У младенцев с врожденными дефектами гипофиза или гипоталамуса в неонатальном периоде могут возникать такие неотложные состояния, как апноэ, цианоз или тяжелые гипогликемии, с судорогами или без них. Нередко встречается длительная неонатальная холестатическая желтуха. Она характеризуется повышением уровней как конъюгированного, так и неконъюгированного билирубина и может быть связана с неонатальным гигантоклеточным гепатитом. Нистагм может указывать на септооптическую дисплазию. Дополнительным диагностическим признаком является микропенис. Дефицит ГР может сопровождаться надпочечниковой недостаточностью, гипотиреозом, а также дефицитом гонадотропинов.
При осмотре обращают на себя внимание круглая голова, короткое и широкое лицо. Лоб выпуклый, переносица вдавленная или седловидная. Нос маленький, с отчетливыми носогубными складками. Нижняя челюсть и подбородок недоразвиты, зубы прорезываются поздно, тесно расположены. Шея и гортань укорочены. Голос высокий и остается таким после пубертата. Телосложение пропорциональное, кисти и стопы маленькие. Вес обычно соответствует росту, но из-за избытка жировой ткани и дефицита мышечной массы малыш выглядит «пухленьким». Размеры половых органов обычно уменьшены, а половое развитие запаздывает или вообще не наступает. Оволосение лица, подмышечных впадин и лобка отсутствует; волосы на голове тонкие. Интеллект обычно соответствует возрасту (если нет других структурных аномалий головного мозга); по сравнению с детьми своего роста пациенты могут казаться не по годам развитыми.
2. Приобретенный гипопитуитаризм. У ранее здорового ребенка в результате полного или почти полного разрушения гипофиза постепенно появляются и прогрессируют признаки идиопатической гипофизарной недостаточности. Атрофия коры надпочечников, ЩЖ и гонад приводит к потере МТ, астении, непереносимости холода, ступору и отсутствию потоотделения. Половое развитие не запускается или регрессирует, если оно уже началось. Атрофия гонад и полового тракта может сопровождаться аменореей, потерей волос на лобке и в подмышечных впадинах. Имеется тенденция к гипогликемии. Рост резко замедляется. Возможно развитие несахарного диабета (см. главу 574), который может быть скрыт центральной надпочечниковой недостаточностью.
Если причиной повреждения гипофиза служит растущая опухоль, то могут появиться такие симптомы, как головная боль, рвота, нарушения зрения и сна, снижение школьной успеваемости, судороги, полиурия, задержка роста. Замедлению темпов роста могут предшествовать неврологические признаки и симптомы, особенно при краниофарингиоме. В некоторых случаях проявления гипофизарной недостаточности впервые возникают после хирургического вмешательства. У детей с краниофарингиомами часто встречаются сужение полей зрения, атрофия зрительного нерва, отек диска зрительного нерва, парез черепных нервов, а также ожирение.
д) Лабораторные признаки. Заподозрить дефицит ГР следует у детей с тяжелой постнатальной задержкой роста (табл. 6). Критериями низкорослости служат рост ниже 1-го процентиля для данного возраста и пола или рост >2 SD ниже среднего значения роста родителей с поправкой на пол. Приобретенный дефицит ГР может возникнуть в любом возрасте, и при его остром начале рост может оказаться в пределах возрастной нормы. Скорость роста пациентов как с врожденным, так и приобретенным дефицитом ГР будет низкой по сравнению со сверстниками того же пола и костного возраста. Для диагностики дефицита ГР важны клинические данные, поскольку результаты лабораторных исследований недостаточно специфичны.
Определенное диагностическое значение имеют низкие сывороточные уровни ИФР-1 и ГР-зависимого ИФР-СБЗ, если сравниваются с нормальными для костного возраста, а не хронологического. Значения ИФР-1 и ГР-зависимого ИФР-СБЗ, близкие к верхней границе нормы для данного возраста, исключают дефицит ГР. Уровень ИФР-1 ближе к нижней границе возрастной нормы может быть у здоровых нормально растущих детей, детей с недостаточностью питания или гипопитуитаризмом. В младенчестве и раннем возрасте ожидаемые пределы значений ИФР-1 у здоровых детей и детей с дефицитом ГР отчасти перекрываются. Использовать для диагностики дефицита ГР исключительно уровни ИФР-1 не рекомендуется.
Долгое время считалось, что окончательный диагноз дефицита ГР требует доказательства отсутствия ГР или снижения его ответа на стимуляцию, однако стимулирующие пробы можно не проводить пациентам с предполагаемыми для дефицита ГР ауксологическими данными, задокументированными дефектами гипоталамуса или гипофиза и недостаточности еще одного, как минимум, гормона гипофиза. Существуют различные провокационные тесты, которые вызывают быстрое повышение уровня ГР у здоровых детей. К ним относятся пробы с введением инсулина, аргинина, клонидина, леводопы или глюкагона. Поскольку гормоны ЩЖ необходимы для нормальной продукции ГР, их уровень должен быть оценен до проведения ГР-стимуляционных проб.
Сниженный линейный рост, отставание костного возраста и низкий уровень пика ГР (<10 нг/мл) в двух из таких проб указывают на хронический дефицит ГР. Об остром дефиците ГР свидетельствуют клинические признаки дефицита ГР и низкий уровень пика ГР (<10 нг/мл) в двух из таких проб. Значение пика ГР <10 нг/мл — довольно условная точка отсечения, она выше, чем таковая для диагностики дефицита ГР у взрослых. Единого мнения относительно критериев дефицита ГР, учитывающих возраст, пол и количественные характеристики ГР, не существует. Некоторые исследования показывают, что у большого числа здоровых детей препубертатного возраста уровень ГР в двух пробах не достигает пика >10 нг/мл. Для повышения диагностической специфичности тестов было предложено предварительное краткосрочное введение половых стероидов.
Помимо установления диагноза дефицита ГР необходимо исследовать др. функции гипофиза. Уровни ТТГ, свободного тироксина или общего тироксина с тестом на поглощение Т3, АКТГ, кортизола, гонадотропинов и половых стероидов могут свидетельствовать о недостаточности др. тропных гормонов. Дефицит АДГ можно установить после проведения соответствующих исследований.
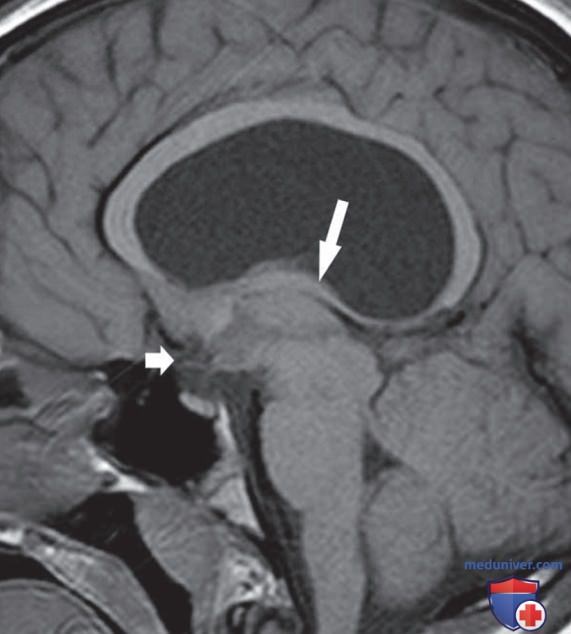
е) Результаты радиологических исследований. Если причина гипопитуитаризма неизвестна, необходима нейровизуализация. КТ подходит для выявления супраселлярных кальцификатов, ассоциированных с краниофарингиомой, а также костных изменений, сопровождающих гистиоцитоз. МРТ дает гораздо более подробное изображение гипоталамо-гипофизарной области. Во многих случаях тяжелого МДГГ с ранним началом обнаруживается триада: гипоплазия передней доли гипофиза, аплазия или гипоплазия гипофизарной ножки и эктопия яркого пятна задней доли гипофиза у основания гипоталамуса (рис. 3). Сниженная высота передней доли гипофиза подразумевает малый размер гипофиза, что часто встречается при генетическом и идиопатическом ИДГР.
Рисунок 3. Сагиттальное Т1-взвешенное магнитно-резонансное изображение демонстрирует эктопию задней доли гипофиза (белая стрелка) и гипоплазию передней доли гипофиза (черная стрелка)
Краниофарингиома является частой причиной приобретенного гипопитуитаризма, в то время как аденомы гипофиза у детей встречаются редко. У пациентов с мутациями генов PROP1 или LHX3 наблюдается как гипоплазия гипофиза, так и его гиперплазия.
Костное созревание можно оценить с помощью обычной рентгенографии кисти (костный возраст), оно замедлено у пациентов с ИДГР; при комбинированном дефиците ГР и ТТГ наблюдается еще более значительная его задержка. Двухфотонная рентгеновская абсорбциометрия показывает снижение минерализации костей, дефицит мышечной массы и соответствующее увеличение жировой массы, однако использовать метод в рутинной практике диагностики дефицита ГР у детей не рекомендуется.
ж) Дифференциальная диагностика. Причины нарушения роста многочисленны: гормональные нарушения, хронические заболевания, недостаточность питания, генетические заболевания, несиндромальные семейные особенности и конституциональная задержка роста и развития. Гормональные нарушения включают гипотиреоз и синдром Кушинга. Из системных заболеваний следует учитывать ВЗК, целиакию, ХБП и анемию. При таких заболеваниях дети часто отстают в МТ больше, чем в росте. Тяжелая психосоциальная депривация может привести к задержке роста, имитирующей дефицит ГР. Первым признаком многих синдромальных генетических заболеваний является низкорослость. Некоторые генетические заболевания, такие как синдром Тернера или дефекты гена SHOX, имеют различные фенотипы, но иногда их единственным клиническим проявлением может стать низкорослость.
В некоторых случаях отставание в росте (рост <2,25 SDS среднего по возрасту) и темпах роста (5 см и менее в год) наблюдается у здоровых детей с нормальной стимулированной и спонтанной секрецией ГР; такое состояние часто называют идиопатическая низкорослость. Уровни ИФР-1 в плазме у таких пациентов могут быть нормальными или сниженными. Лечение таких детей ГР в дозах, применяемых при гипопитуитаризме, в большинстве случаев улучшает их антропометрические показатели, а конечный рост, по данным различных исследований, составляет от -2,5 до +7,5 см по сравнению с прогнозируемым. Заранее прогнозировать, у кого из получающих ГР детей конечный рост будет выше расчетного, а у кого ниже, невозможно.
Стратегии ДД постоянного ДГР и др. форм низкорослости несовершенны. При сочетании генетической низкорослости и конституциональной задержки роста дети имеют низкий рост, отставание костного возраста и, во многих случаях, минимальный выброс ГР при проведении провокационных проб. Когда взрослые пациенты с диагностированным в детстве идиопатическим или приобретенным дефицитом ГР и получавшие чГР проходят ретестирование, у большинства из них уровни пика ГР находятся в пределах нормальных значений.
1. Конституциональная задержка роста. Педиатры часто встречаются с конституциональной задержкой роста, являющейся одним из вариантов нормального развития. Длина и МТ при рождении у таких детей в норме, так же как и рост в первые 4-12 мес жизни. В детстве показатели роста находятся на более низком процентиле. Поскольку половое развитие задерживается, темпы роста продолжают снижаться, в то время как у сверстников начинается пубертатное ускорение роста. Путем тщательного опроса удается обычно выявить аналогичную динамику роста и полового развития в анамнезе одного или обоих родителей, имеющих нормальный конечный рост. Уровень ИФР-1, как правило, снижен для хронологического возраста, но нормальный для костного возраста. Ответ ГР на провокационные пробы обычно ниже, чем у детей с более своевременным пубертатом.
Прогноз для достижения такими детьми нормального конечного роста благоприятный. Прогнозы для конечного роста, основанные на росте и костном возрасте, могут быть завышены, и скорее у мальчиков, чем у девочек. В отношении мальчиков с задержкой полового созревания >2 лет короткий курс терапии тестостероном может способствовать ускорению полового развития после 14-летнего возраста. Считается, что причиной конституциональной задержки роста является сохранение относительно низкого уровня гонадотропинов в детстве.
з) Лечение. Рекомбинантный чГР (рчГР) отпускаетсся по рецепту с 80-х гг. прошлого столетия. В США представлено несколько брендов. Это одинаковые с терапевтической точки зрения препараты, основные различия которых заключаются в запатентованных устройствах для подкожных инъекций и доступности растворимых жидких форм по сравнению с порошками, которые необходимо разводить перед инъекцией. Пролонгированных форм в настоящее время нет; клинические испытания таких ЛП продолжаются.
FDA одобрило восемь показаний для лечения детей рчГР для улучшения линейного роста. Это дефицит ГР, синдром Тернера, ХБП (перед трансплантацией), идиопатическая низкорослость, внутриутробная задержка роста, синдром Прадера-Вилли, мутации гена SHOX и синдром Нунан. Одобрение лечения данных заболеваний FDA не гарантирует, что страховая компания одобрит оплату за ЛП. Лечение следует начинать как можно раньше, чтобы сократить отставание в росте от одноклассников в детстве и добиться максимального влияния на конечный рост. Рекомендуемая начальная доза рчГР для лечения дефицита ГР составляет 0,16-0,24 мг/кг/нед (22-35 мкг/кг/сут) (в РФ 0,033 мг/кг/сут*).
P.S. * Федеральные клинические рекомендации (протоколы) по ведению детей с эндокринными заболеваниями. Под ред. И.И. Дедова и В.А. Петерковой. М.: Практика, 2014. С. 360.
Более высокие дозы используются в период полового развития и по показаниям, не связанным с дефицитом ГР. РчГР вводится п/к 1 р/сут Максимальный ответ на терапию рчГР наблюдается в первый год лечения. Скорость роста в первый год терапии обычно превышает 95-й процентиль для соответствующего возраста. С каждым последующим годом лечения скорость роста снижается. Если скорость роста падает <25-го процентиля, следует оценить соблюдение режима инъекций, прежде чем увеличивать дозу. Уровень ИФР-1 можно использовать в качестве объективной оценки соблюдения режима лечения. Терапию ГР следует продолжать до достижения пациентом роста, близкого к конечному. Критериями прекращения лечения ГР являются: решение пациента о том, что он или она достаточно высок(а), скорость роста <2,5 см/год, костный возраст >14 лет у девочек и >16 лет у мальчиков.
Одновременное лечение рчГР и агонистами ГнРГ использовалось в надежде остановить половое развитие, задержать закрытие эпифизарных зон роста и продлить период роста. Такая стратегия способна улучшить прогноз для конечного роста, но одновременно может увеличить разрыв в созревании между пациентами и их сверстниками, а также ухудшить минерализацию костей. Предпринимались попытки предотвратить закрытие эпифизарных зон роста у мальчиков с помощью терапии ингибиторами ароматазы, которые блокируют фермент, ответственный за превращение андрогенов в эстрогены; клинические испытания для оценки эффективности такого подхода продолжаются. У некоторых пациентов на фоне терапии ГР обнаруживается первичный или центральный гипотиреоз.
Существует также риск развития надпочечниковой недостаточности как компонента гипопитуитаризма. Нераспознанная, она может привести к летальному исходу. Всем пациентам с диагностированным дефицитом ГР показана периодическая оценка функции ЩЖ и надпочечников.
Лечение рчГР может ускорить рост детей и без дефицита ГР. В настоящее время проводится интенсивное исследование с целью определить весь спектр детей с низкорослостью, которым лечение ГР может помочь. FDA одобрило использование ГР при идиопатической низкорослости при росте <1,2 процентиля (-2,25 SD) для соответствующего возраста и пола, прогнозируемом росте <5-го процентиля и открытых зонах роста. Исследования влияния лечения ГР на конечный рост показывают, что средний прирост составляет 5-7,6 см в зависимости от общей дозы и длительности лечения.
У детей с МДГГ заместительная терапия должна быть направлена на компенсацию недостаточности и др. гормонов гипофиза. При недостаточности ТТГ назначаются тиреоидные гормоны в полной заместительной дозе. При дефиците АКТГ рекомендуется гидрокортизон в физиологических дозах, 8-12 мг/м2/сут.
Примечание. Согласно КР РФ, доза должна быть по возможности приближена к наименьшей заместительной дозе 5-10 мг/м2/сут, максД 12 мг/м2/сут*.
P.S. * Федеральные клинические рекомендации (протоколы) по ведению детей с эндокринными заболеваниями. Под ред. И.И. Дедова и В.А. Петерковой. М.: Практика, 2014. С. 364.
Индивидуальный подбор дозы должен сводить к минимуму риск побочных эффектов от избыточного поступления глюкокортикоидов и предотвращать симптомы надпочечниковой недостаточности. Во время заболеваний и перед оперативными вмешательствами дозы гидрокортизона увеличивают. Пациентам с дефицитом гонадотропинов половые стероиды назначают, когда костный возраст достигает возраста, соответствующего началу полового развития. У младенцев с микропенисом один или два 3-месячных курса ежемесячных инъекций 25 мг тестостерона ципионата или тестостерона энантата могут нормализовать размер полового члена, не слишком влияя на созревание скелета.
При первичном дефиците ИФР-1 одобрено на территории США применение рекомбинантного ИФР-1 (мекасермин) п/к 2 р/сут. Побочные эффекты аналогичны рчГР, за исключением того, что мекасермин может вызывать гипогликемию. Риск гипогликемии снижается, если инъекции делать во время еды или перекуса. В таких случаях, как дефекты генов рецептора ГР и STAT5b (изменяющие последующую передачу сигналов ГР), использование мекасермина более эффективно, чем ГР. Также ЛП м.б. полезен у редких пациентов с тяжелым дефицитом ГР и высокими уровнями АТл к экзогенному рчГР. Однако мекасермин не показан для лечения большинства пациентов с ДГР.
и) Осложнения и побочные эффекты терапии гормоном роста. Лечение ГР влияет на гомеостаз глюкозы. Уровни инсулина натощак и после еды обычно низкие до лечения, нормализуются на фоне заместительной терапии ГР. Лечение ГР ассоциировано с увеличением риска развития СД-2; значительного увеличения риска развития СД-1 не отмечается. Высказывались опасения по поводу безопасности применения ГР у детей, дефицит ГР у которых развился после лечения опухолей ГМ, лейкемии и др. новообразований. Долгосрочные исследования не свидетельствуют об увеличении риска рецидива краниофарингиомы, др. опухолей ГМ или лейкемии. Как минимум 3 исследования указывают на повышенный риск вторичных новообразований у пациентов, перенесших рак и получавших терапию ГР.
Неподтвержденное исследование свидетельствует о повышенном риске геморрагического инсульта и 30% увеличении смертности среди молодых людей, получавших ГР в детстве, особенно если доза ГР превышала 0,35 мг/кг/нед (50 мкг/кг/сут). В др. исследовании описываются такие побочные эффекты, как псевдоопухоль ГМ, смещение эпифиза головки бедренной кости, гинекомастия, укрупнение черт и прогрессирование сколиоза. Риск позднего развития болезни Кройтцфельдта-Якоба был ограничен получателями загрязненных партий экстрагированного гипофизарного ГР. Использование рчГР, который является единственной лекарственной формой чГР, используемой в настоящее время в клинической практике, исключает подобный риск.