Онкологические заболевания — это группа заболеваний, возникающих в результате альтераций, происходящих в самых разных генах. Для того чтобы клетки стали в полной мере злокачественными, требуются множественные мутации, некоторые из которых являются герминальными, но большинство из них приобретенные (соматические). Эти мутации приводят к альтерациям в нормальных клеточных процессах, контролирующих пролиферацию и выживание клеток, включая сигнальную трансдукцию, контроль клеточного цикла, репарацию ДНК, клеточный рост и дифференцировку, трансляционную регуляцию, старение и апоптоз (запрограммированную гибель клеток).
а) Гены, участвующие в онкогенезе. В развитие онкологических заболеваний вовлечены два основных класса генов: онкогены и гены-супрессоры опухолей. Протоонкогены — это клеточные гены, имеющие значение для нормального функционирования клеток и кодирующие разл. белки, включая факторы транскрипции, факторы роста и рецепторы факторов роста. Эти белки являются жизненно важными компонентами в сети сигнальной трансдукции, они регулируют рост, деление и дифференцировку клеток.
Протоонкогены могут подвергаться альтерациям и превращаться в онкогены, т.е. гены, которые, будучи транслированы, могут вызвать злокачественную трансформацию клетки.
Онкогены можно разделить на пять разл. классов в зависимости от механизмов их действия. Изменения в любом из нормальных клеточных компонентов могут привести к неконтролируемому росту клеток. Некоторые онкогены кодируют факторы роста, связывающиеся с рецептором, и стимулируют выработку белка. Др. онкогены кодируют рецепторы фактора роста — белки, находящиеся на поверхности клеток. Когда факторы роста связываются с рецептором фактора роста, они могут «включать»/«выключать» этот рецептор.
Мутационные/посттрансляционные модификации рецептора могут привести к постоянному включению рецептора с последующим нерегулируемым ростом клетки. Еще один класс представлен сигнальными трансдукторами/эффекторами. Сигнальные трансдукторы отвечают за передачу сигнала от рецептора на клеточной поверхности к ядру клетки. Факторы транскрипции — это молекулы, которые связываются с определенными участками ДНК и контролируют транскрипцию. МУС и MYCN являются примерами факторов транскрипции, которые при активации мутацией/амплификацией начинают чрезмерно стимулировать клеточное деление.
Последний класс онкогенов препятствует апоптозу. В результате того, что клетки перестают реагировать на сигнал к разрушению, происходит неконтролируемая клеточная пролиферация.
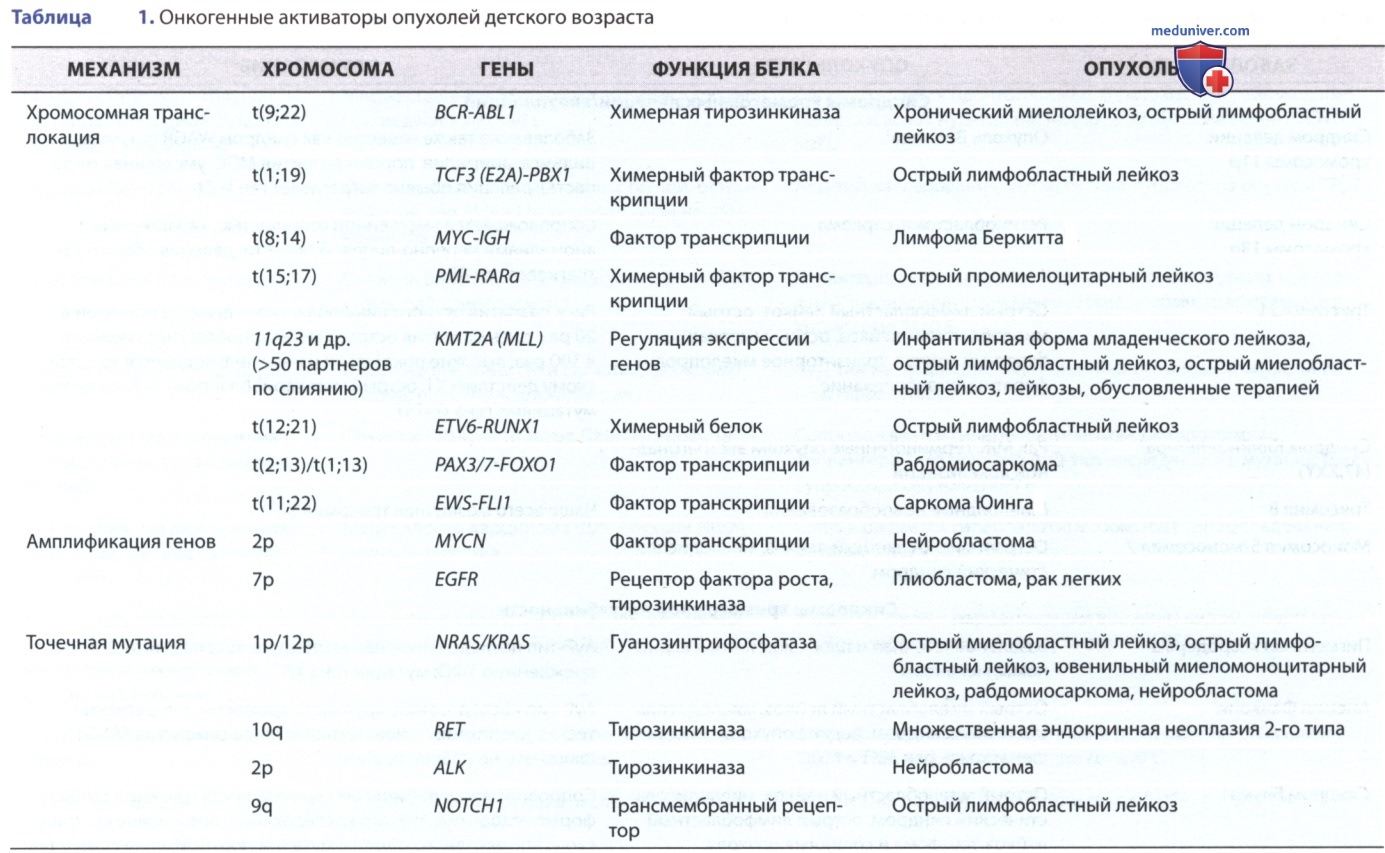
Протоонкогены активируются 3 основными механизмами, как то: амплификация, мутация и транслокация/интерстициальная делеция (табл. 1). MYC/MYCN, которые кодируют белки, регулирующие транскрипцию, являются примерами протоонкогенов, активируемых амплификацией. Пациенты с нейробластомой, у которых ген MYCN амплифицирован в 10-300 раз, имеют худший клинический прогноз. Точечные мутации также могут активировать протоонкогены.
Протоонкоген NOTCH1 кодирует мембраносвязанный рецептор, который имеет важнейшее значение для клеточной судьбы и путей дифференцировки во время нормального развития и который подвергается протеолитическому расщеплению при активации, вызванной лигандом, так что белок может проникнуть в ядро клетки и активировать транскрипцию целевого гена. Мутация гена NOTCH1 наблюдается по крайней мере в 50% случаев Т-клеточного острого лимфобластного лейкоза, что приводит к конститутивной активации белка, играющего важную роль в лейкемогенезе.
Хромосомная транслокация/интерстициальная делеция является третьим механизмом, с помощью которого активируются протоонкогены. При некоторых лейкозах и лимфомах последовательности, контролирующие факторы транскрипции, смещаются перед Т-клеточными рецепторами/генами Ig, что приводит к нарушению регуляции транскрипции этих генов и лейкемогенезу. Ярким примером являются транслокации, вызывающие попадание c-MYC под контроль гена тяжелой цепи Ig (IGH)/генов легкой цепи типа каппа (IGκ)/лямбда (IGλ) при лимфоме Беркитта.
Хромосомные транслокации, соединяющие гены из 2 разных хромосом/вызывающие интерстициальные делеции/инверсии внутри хромосомы, также могут привести к слиянию генов; в результате транскрипции гибридных генов происходит образование химерного белка с новой и потенциально онкогенной активностью. Примерами онкологических заболеваний, вызванных гибридными генами, являются солидные опухоли детского возраста — саркома Юинга [t(11;22)] и альвеолярная рабдомиосаркома [t(2;13)/t(1;13)].
Эти транслокации приводят к появлению новых транскриптов матричной РНК, которые м.б. использованы в качестве диагностических маркеров. Наиболее хорошо описанной транслокацией при лейкозе является Филадельфийская хромосома t(9;22), которая продуцирует белок BCR-ABL1, обнаруживаемый при хроническом миелолейкозе и специфических подтипах острого лимфобластного лейкоза. BCR-ABL1 является конститутивно активной тирозинкиназой. Кроме того, белок локализуется в цитоплазме вместо ядра, подвергая киназу воздействию нового спектра субстратов.
Нарушение регуляции генов-супрессоров опухолей представляет собой еще один механизм, участвующий в онкогенезе. Гены-супрессоры опухолей играют важную роль в регуляции клеточного роста и апоптоза. Их называют рецессивными онкогенами, поскольку для экспрессии злокачественного фенотипа требуется инактивация обоих аллелей гена-супрессора опухоли.
Двухударная теория онкогенеза Кнудсона (Knudson) была разработана на основе ретинобластомы — ЗНО сетчатки глаза, развивающегося в значительно более раннем возрасте у детей с семейной формой заболевания по сравнению со спорадической; такие опухоли часто были мультифокальными при семейных формах, но почти всегда — монофокальными в спорадических случаях.
Кнудсон утверждал, что спорадические ретинобластомы обусловлены соматическими мутациями, инактивирующими обе копии гена, тогда как при семейной ретинобластоме дети наследуют инактивированный аллель от одного родителя и, следовательно, для развития семейной ретинобластомы необходима соматическая инактивация только одного нормального аллеля. Эта гипотеза была доказана 15 лет спустя после открытия гена-супрессора опухоли RB.
Др. важным белком-супрессором опухоли является ТР53, который известен как «страж генома», т.к. он обнаруживает наличие хромосомных повреждений и предотвращает деление клетки до момента репарации. При наличии повреждения, не подлежащего репарации, ТР53 инициирует апоптоз, и клетка умирает. Мутантные белки ТР53 обнаруживаются в клетках >50% ЗНО. Мутации в гене этого белка могут способствовать развитию многих видов ЗНО, включая рак МЖ, колоректальный рак, рак легких, пищевода, желудка, яичников и предстательной железы, а также глиомы, саркомы и некоторые лейкозы.
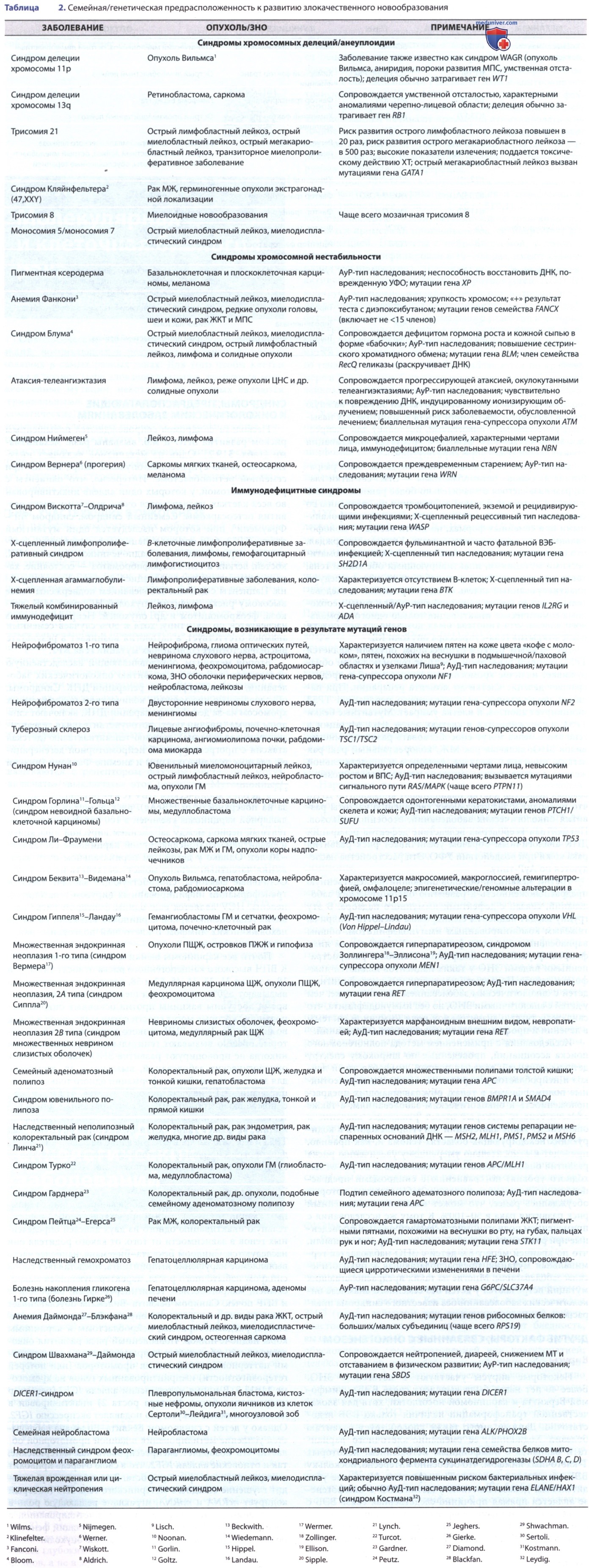
б) Синдромы, предрасполагающие к онкологическим заболеваниям. Несколько синдромов сопровождаются повышенным риском развития ЗНО и м.б. вызваны разл. механизмами (табл. 2). Один из механизмов включает инактивацию генов-супрессоров опухоли, напр., ген RB при семейной ретинобластоме. Интересно, что пациенты с ретинобластомой, у которых один аллель инактивирован во всех клетках, подвергаются очень высокому риску развития остеосаркомы.
Семейный синдром, синдром Ли-Фраумени (Li, Fraumeni), при котором наследуется один мутантный аллель ТР53, был описан у пациентов с саркомами, лейкозами, карциномой коры надпочечников и раком МЖ, костей, легких и ГМ. Нейрофиброматоз — состояние, характеризующееся пролиферацией клеток нервного гребня. Пациенты с данным заболеванием подвержены более высокому риску развития опухолей НС, рака МЖ, лейкоза, феохромоцитом и др. опухолей.
Нейрофиброматоз наследуется по АуД-типу, хотя в 50% случаев семейный анамнез отсутствует, а патология возникает в результате высокочастотных спонтанных мутаций гена NF1.
Второй механизм, обусловливающий наследственную предрасположенность к развитию онкологических заболеваний, связан с дефектами репарации ДНК. Синдромы, обусловленные чрезмерным количеством поврежденных хромосом из-за дефектов репарации ДНК, включают синдром Блума (низкий рост, фоточувствительная телеангиэктатическая эритема), атаксию-телеангиэктазию (детская атаксия с прогрессирующей нейромоторной дегенерацией, глазные телеангиэктазии) и анемию Фанкони (низкий рост, дефекты развития скелета и почек, панцитопения).
В результате снижения способности к репарации хромосомных дефектов клетки накапливают аномальную ДНК, что приводит к значительному увеличению частоты развития онкологических заболеваний, особенно лейкозов. Пигментная ксеродерма вследствие дефектов репарации ДНК аналогичным образом увеличивает риск развития рака кожи при воздействии УФО. Эти расстройства наследуются по АуР-типу.
Третий механизм, обусловливающий наследственную предрасположенность к развитию онкологических заболеваний, связан с дефектами иммунного надзора. В эту группу входят пациенты с синдромом Вискотта-Олдрича, тяжелым комбинированным иммунодефицитом, общим вариабельным иммунодефицитом и Х-сцепленным лимфопролиферативным синдромом. Наиболее распространенными видами ЗНО у таких пациентов являются лимфома и лейкоз.
Показатели излечения иммунодефицитных детей с онкологическим заболеванием намного ниже, чем у детей с аналогичными ЗНО, но без иммунодефицита, что свидетельствует о значительной роли иммунной системы в лечении и профилактике онкологических заболеваний.
Исследования с применением метода полногеномного поиска ассоциаций, проведенные по широкому спектру детских опухолей, включая острый лимфобластный лейкоз и нейробластому, определили общие однонуклеотидные полиморфизмы в генах, обуславливающих предрасположенность к онкологическим заболеваниям, а также области генома, которые имеют решающее значение в онкогенезе.
Такие альтерации могут происходить в кодирующих/некодирующих областях генома и, как правило, приводят к относительно умеренному увеличению риска развития онкологического заболевания (в 2-10 раз от исходного уровня) по сравнению с синдромами предрасположенности к онкологическим заболеваниям, которые обсуждались ранее, что может вызывать пожизненный риск развития рака в 50-100%. К тому же исследования с применением полногеномного секвенирования, проведенные при разл. видах опухолей детского возраста, выявили, что по крайней мере у 8% детей с ЗНО наблюдается герминальная мутация, предрасполагающая к онкологическим заболеваниям.
Многие из таких предрасполагающих мутаций встречаются у детей без семейного анамнеза онкологических заболеваний/без известного синдрома предрасположенности к развитию рака.
в) Другие факторы, связанные с онкогенезом:
1. Вирусы. Некоторые вирусы участвуют в патогенезе ЗНО. Более 40 лет назад была выявлена связь ВЭБ с лимфомой Беркитта и карциномой носоглотки, хотя для злокачественной трансформации наличия только ВЭБ недостаточно. Данный вирус также способствует развитию болезни Ходжкина (Hodgkin), ее смешанно-клеточного варианта и варианта с лимфоидным истощением, некоторых Т-клеточных лимфом, что особенно интересно, поскольку ВЭБ обычно не поражает Т-лимфоциты.
Наиболее убедительным доказательством участия ВЭБ в лимфогенезе является прямая причинно-следственная связь ВЭБ с В-клеточным лимфопролиферативным заболеванием у пациентов с ослабленным иммунитетом, особенно у пациентов с ВИЧ-инфекцией/находящихся на иммуносупрессивной терапии после трансплантации органов. ВГЧ 8 типа способствует развитию саркомы Капоши (Kaposi).
У детей с хроническим HBV (с «+» результатом анализа на поверхностный HBeAg), риск развития гепатоцеллюлярной карциномы увеличен в 100 раз. У взрослых латентный период между заражением вирусной инфекцией и развитием гепатоцеллюлярной карциномы составляет ~20 лет. Однако у детей при перинатальном пути заражения латентный период может составлять не >6-7 лет. Остальные факторы, способствующие злокачественной трансформации инфицированных вирусом гепатоцитов, неясны.
HCV является еще одним фактором риска развития гепатоцеллюлярной карциномы, вызывает развитие неходжкинских лимфом В-клеточной подгруппы, напр., лимфомы селезенки.
Почти все карциномы шейки матки вызываются ВПЧ. К ВПЧ высокого канцерогенного риска относят типы 16 и 18, а также 31, 33, 34, 45, 52 и 58, которые в совокупности вызывают >90% случаев рака шейки матки. В настоящее время доступны вакцины против основных онкогенных подтипов, которые могут спасти сотни миллионов жизней во всем мире. Типы ВПЧ низкого риска, в т.ч. 6 и 11, которые обычно вызывают генитальные бородавки, почти никогда не провоцируют развитие ЗНО. Как и в случаях онкологических заболеваний, вызванных др. вирусами, для злокачественной трансформации одного только инфицирования ВПЧ недостаточно.
Считается, что механизм, с помощью которого ВПЧ-ассоциированные онкопротеины Е6 и Е7 индуцируют злокачественную трансформацию, включает такие белки-супрессоры опухолей, как ТР53 и RB, а также др. пути, имеющие решающее значение для прогрессирования клеточного цикла, поддержания стабильности теломеразы и генома, апоптоза.
2. Геномный импринтинг. Развитие онкологических заболеваний связано с геномным импринтингом, который представляет собой избирательную инактивацию одного из двух аллелей определенных генов в зависимости от того, от какого родителя они наследуются. Синдром Беквита-Видемана, наиболее часто выявляемое нарушение импринтинга, представляет собой синдром избыточного роста, характеризующийся макросомией, макроглоссией, гемигипертрофией, омфалоцеле и ВПР почек. Синдром Беквита-Видемана обуславливает повышенный риск развития опухоли Вильмса, гепатобластомы, рабдомиосаркомы, нейробластомы и карциномы коры надпочечников.
Повышенный риск развития онкологического заболевания напрямую связан с изменениями паттернов метилирования промоторов (или потерей гетерозиготности) импринтированных генов на хромосоме 11р15.5. В норме материнский аллель IGF2 (рецептор инсулиноподобного фактора роста 2) инактивирован в данном геномном локусе, что подавляет экспрессию IGF2. Однако у детей с синдромом Беквита-Видемана наблюдается усиление метилирования в данной промоторной области, что позволяет экспрессировать как материнские, так и отцовские аллели IGF2, что в свою очередь приводит к сверхэкспрессии фактора роста.
В то же время происходит глушение соседнего материнского гена Н19 (который кодирует ncRNA и miRNA, играющие решающую роль в подавлении роста) посредством гиперметилирования, в конечном итоге формируются соответствующий фенотип роста и предрасположенность к развитию опухоли.
3. Теломераза. Теломеры представляют собой серию из десятков/тысяч повторяющихся участков TTAGGG на концах хромосом. Теломеры участвуют в стабилизации концов хромосом и ограничении процессов разрушения, транслокации и потери материала ДНК. При репликации ДНК происходит прогрессирующее укорочение длины теломер, что является признаком клеточного старения. Этот процесс также называется репликативным старением. При большинстве видов онкологических заболеваний из-за мутации в промоторной области гена TERT происходит активация теломеразы (кодируемой геном TERT) — фермента, добавляющего теломеры к концам хромосом.
Поддержание длины теломер в опухолевых клетках с помощью теломеразы способствует снятию барьеров, контролирующих жизненный цикл клеток, что приводит к неограниченной клеточной пролиферации.