P.S. На момент подготовки данной статьи клинических рекомендаций МЗ РФ по данной тематике нет.
Аутоиммунный полигландулярный синдром (АПС) возникает, когда аутоиммунная активность направлена на несколько эндокринных желез и/или неэндокринные органы, иногда в сочетании с иммунодефицитом. Эндокринные железы и др. органы, чаще всего поражаемые при АПС, имеют специфические аутоАГн, повышающие восприимчивость этих тканей к повреждению неконтролируемым иммунным ответом.
Большинство аутоиммунных эндокринопатий обусловлены клеточно-опосредованным иммунным ответом аутореактивных Т-клеток. Несмотря на то, что АТл к одному/нескольким аутоАГн обычно сочетаются со специфическими аутоиммунными эндокринопатиями, в большинстве случаев они не являются непосредственными патогенами, а скорее служат маркерами иммунной дисрегуляции. Известным исключением является болезнь Грейвса (Graves), вызываемая аутоАТл, непосредственно активирующими рецептор ТТГ.
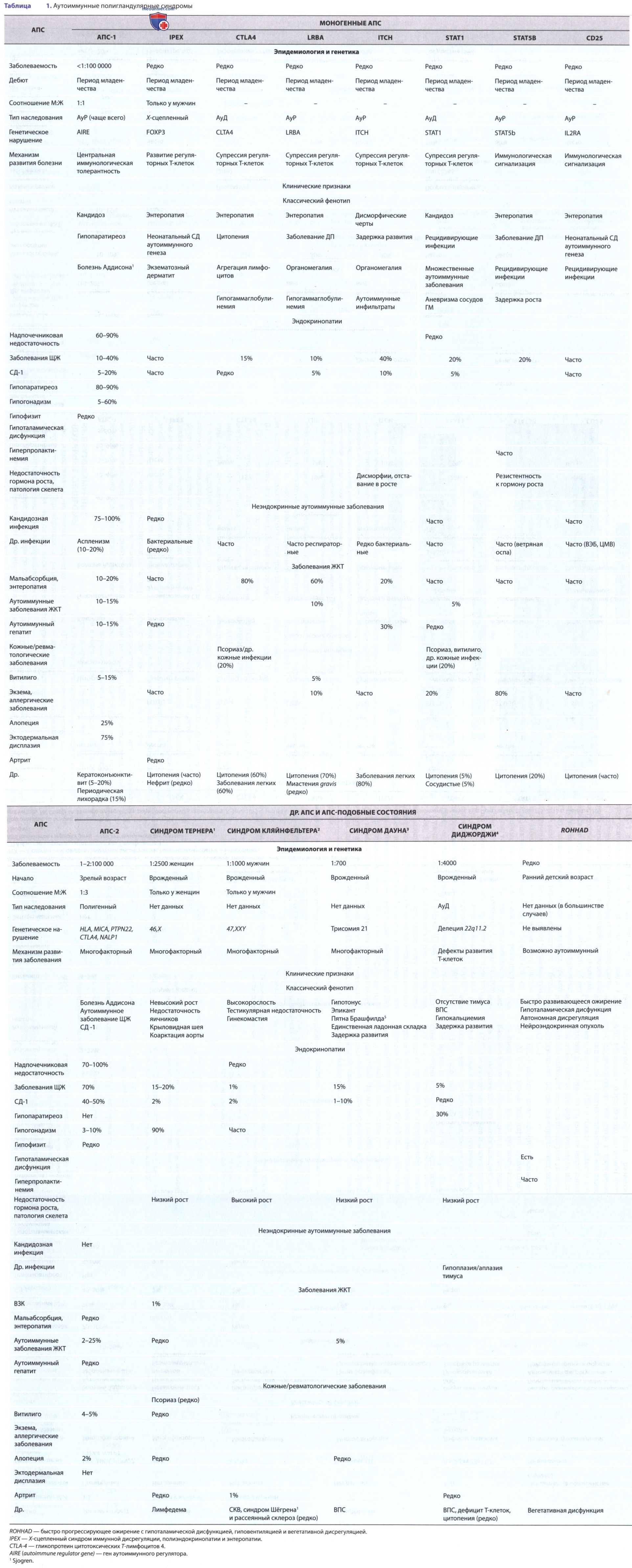
АПС, обусловленный моногенными нарушениями, вызывающими иммунную дисрегуляцию, включая АПС 1-го типа (АПС-1), носит наследственный характер в части ключевых моментов иммунотолерантности (табл. 1). Полигенные нарушения, вызывающие развитие АПС (АПС 2-го типа), и некоторые хромосомные аномалии (напр., трисомия 21), также вызывают аберрантный иммунный ответ, приводящий к полиорганным аутоиммунным расстройствам.
И наконец, негенетические факторы (напр., ингибиторы контрольных точек иммунного ответа на терапию рака) могут приводить к аутоиммунному полиэндокринному заболеванию. Несмотря на то, что АПС встречается редко, у пациентов наблюдается тяжелая хроническая патология, особенно если этот синдром не выявлен на раннем этапе и не лечился надлежащим образом. Между первыми и последующими проявлениями эндокринопатий может пройти 1-2 десятилетия.
Наличие первичного гипопаратиреоза, первичной недостаточности надпочечников, неонатального СД-1, хронического кандидоза кожи и слизистых либо соответствующего семейного анамнеза должно вызывать подозрение на АПС.
а) Моногенные аутоиммунные полигландулярные синдромы. Количество известных моногенных дефектов механизмов иммунорегуляции, приводящих к развитию АПС, существенно выросло за последнее десятилетие (см. табл. 1). Наиболее хорошо охарактеризованные моногенные формы АПС обусловлены в первую очередь мутациями, влияющими на механизмы формирования центральной иммунологической толерантности (АПС-1)/развитие регуляторных Т-клеток [Х-сцепленный синдром иммунной дисрегуляции, полиэндокринопатии и энтеропатии (IPEX)]. Др. моногенные АПС (так называемые IPEX-подобные заболевания) обусловлены дефектами в регуляции Т-клеточной супрессии/сигнализации).
б) Аутоиммунный полигланудлярный синдром первого типа. АПС-1, типичный моногенный полиэндокринный синдром, обусловлен мутациями в гене аутоиммунного регуляторного фактора (AIRE) на хромосоме 21q22.3. AIRE играет решающую роль в презентации аутоАГн формирующимся в тимусе Т-клеткам, что обычно приводит к развитию центральной иммунологической толерантности, индуцируя апоптоз Т-клеток, специфичных для этих аутоАГн (негативная селекция). AIRE также играет определенную роль в формировании регуляторных Т-клеток. Т.о., у пациентов с АПС-1 развиваются аутореактивные Т-клетки и аутоАТл, направленные на многие ткани.
АПС-1 является редким заболеванием, однако чаще встречается в некоторых популяциях коренных народов, включая иранских евреев, сардинцев, финнов и норвежцев, при этом его распространенность колеблется в пределах 1:9000-90 000 человек. Он наследуется по АуР-типу, хотя известны спорадические и АуД-варианты. АПС-1 также имеет клиническое описательное название — аутоиммунная полиэндокринопатия, кандидоз-эктодермальная дистрофия (APECED*).
АПС-1 диагностируют по наличию минимум двух из трех основных клинических признаков (триада Уитакера (Whitaker)): хронического кожно-слизистого кандидоза, первичного гипопаратиреоза и первичной надпочечниковой недостаточности. Три этих заболевания, как правило, развиваются со временем — кандидоз в возрасте <5 лет, гипопаратиреоз ок. 10 лет, а недостаточность надпочечников в ~15 лет, однако точный порядок и возраст проявления каждого компонента значительно варьируют. У большинства пациентов со временем развиваются новые аутоиммунные заболевания, при этом кожные и желудочно-кишечные, как правило, появляются в возрасте <20 лет, а др. эндокринные расстройства — после второго десятилетия жизни (см. табл. 1).
Половая принадлежность, происхождение и специфические мутации AIRE могут коррелировать с повышенным риском определенных проявлений.
При АПС-1 почти каждая эндокринная железа м.б. поражена вследствие нарушения иммунной регуляции. Чаще всего поражаются ПЩЖ и надпочечники (в 80% случаев), реже — гонады (яичники у 70% женщин; тестикулы у 30% мужчин), ЩЖ (20%), β-клетки ПЖЖ (10%) и гипофиз (<5%). Неэндокринные аутоиммунные заболевания, затрагивающие многие ткани организма, могут появляться до первых клинических проявлений эндокринопатии. Чаще всего поражаются зубы и ногти, у большинства пациентов наблюдается эктодермальная дистрофия (80%). Среди др. поражаемых тканей ЖКТ (по 15% случаев приходится на мальабсорбцию, аутоиммунный гепатит и пернициозную анемию), кожа (витилиго у 15%) и волосяные луковицы (алопеция в 25% случаев).
Пациенты с АПС-1 подвержены повышенному риску развития инфекций, что, возможно, обусловлено наличием цитокиновых аутоАТл, дисфункции селезенки и нарушением целостности кишечника. Кожно-слизистый кандидоз встречается очень часто (70-100%) и может привести к развитию рака пищевода в случае отсутствии своевременного выявления и лечения. Рак пищевода, аутоиммунный гепатит, острая надпочечниковая недостаточность и тяжелая гипокальциемия являются основными причинами смерти у пациентов с АПС-1.
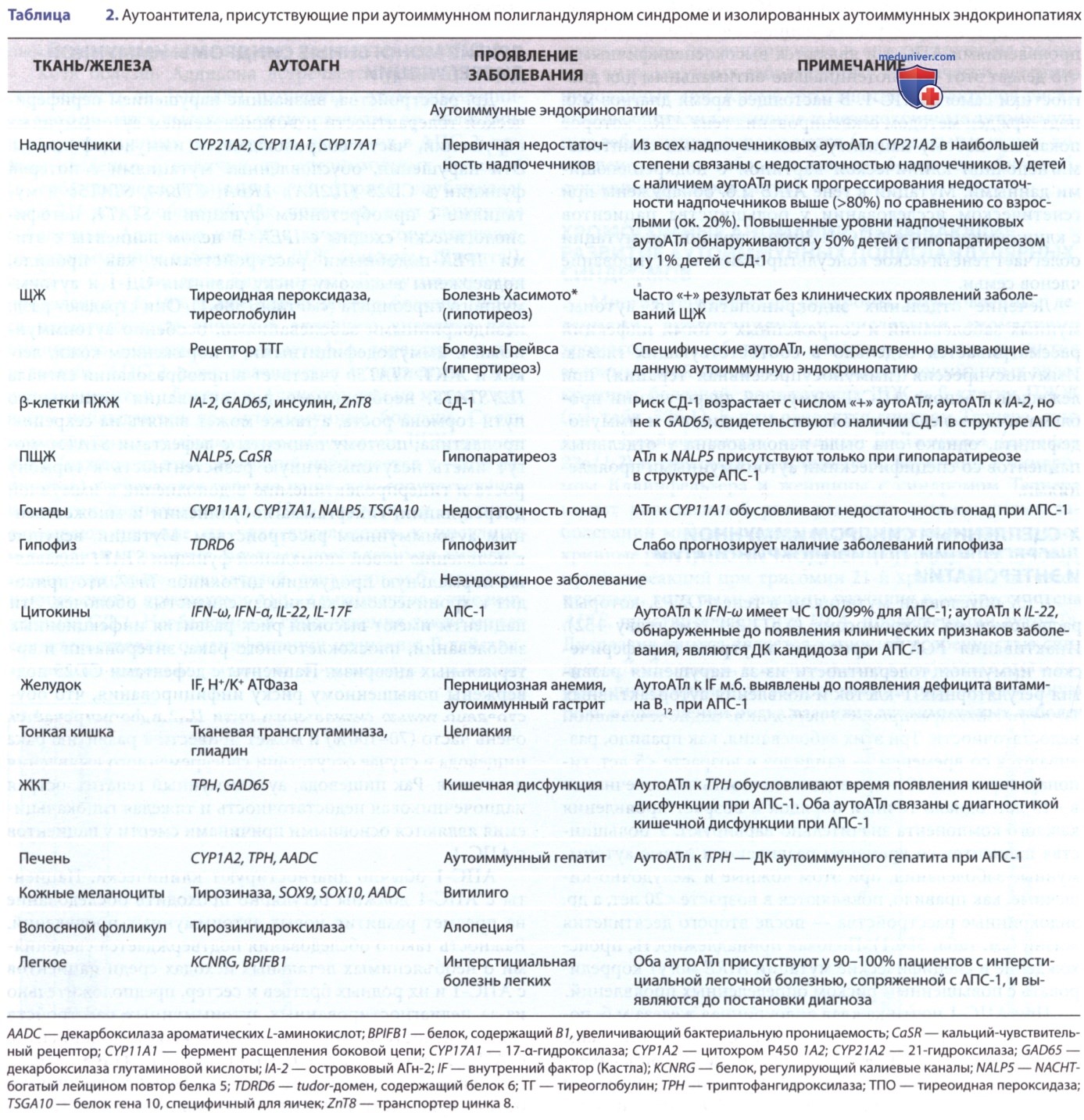
АПС-1 обычно диагностируют клинически. Пациенты с АПС-1 должны регулярно проходить обследование на предмет развития новых аутоиммунных проявлений. Важность такого обследования подтверждается сведениями о необъяснимых летальных исходах среди пациентов с АПС-1 и их родных братьев и сестер, предположительно из-за недиагностированных аутоиммунных расстройств (напр., недостаточности надпочечников). У пациентов с АПС-1 м.б. обнаружены разл. аутоАТл (табл. 2). В то время как некоторые из них присутствуют при соответствующей аутоиммунной эндокринопатии (напр., АТл к 21-гидроксилазе при надпочечниковой недостаточности), др. аутоАТл специфичны только для АПС-1.
Кроме того, клиническая значимость определения специфических для органов аутоАТл для прогнозирования начала эндокринной недостаточности желез при АПС-1 является вариабельной, поэтому клиническое подозрение, лабораторный скрининг и информирование о симптомах развивающихся эндокринопатий и др. аутоиммунных заболеваний имеют первостепенное значение независимо от наличия аутоАТл.
Три вида аутоАТл в итоге могут оказаться диагностически значимыми при АПС-1, хотя они пока не используются в клинической практике. АутоАТл к NALP5 присутствуют при гипопаратиреозе только у пациентов с АПС-1, а не при изолированном аутоиммунном/идиопатическом гипопаратиреозе, в связи с этим пациенты с гипопаратиреозом и АТл к NALP5 должны быть дополнительно обследованы на АПС-1. АутоАТл к цитокинам Th17 (особенно к IL-22 и IL-17F) коррелируют с АПС-1-ассоциированным кандидозом и могут играть важную патогенетическую роль.
АутоАТл к IFN (особенно к IFN-ω и IFN-α) присутствуют практически у всех пациентов с клиническими проявлениями АПС-1 и являются высокоспецифичными, что делает этот тест потенциально оптимальным для диагностики самого АПС-1. В настоящее время диагноз м.б. подтвержден методом секвенирования гена AIRE, которое показано любому пациенту с классическими симптома-ми/неполной клинической картиной с подкрепляющими данными. Мутации в гене AIRE м.б. обнаружены при генетическом исследовании у большинства пациентов с клинически явным АПС-1. Знание причинной мутации облегчает генетическое консультирование и тестирование членов семьи.
Лечение отдельных эндокринопатий, др. аутоиммунных заболеваний и сопряженных с ними инфекций рассматривается отдельно в соответствующих главах. Иммуносупрессия (иммуносупрессивная терапия) при лежащей в основе АПС-1 иммунной дисрегуляции проблематична вследствие сочетания кандидоза и иммунодефицита, однако она была использована у отдельных пациентов со специфическими аутоиммунными проявлениями.
в) Х-сцепленный синдром иммунной дисрегуляции, полиэндокринопатии и энтеропатии. IPEX обусловлен мутациями в гене FOXP3, который расположен на Х-хромосоме (Хр11.23). Инактивация FOXP3 приводит к утрате периферической иммунной толерантности из-за нарушения развития регуляторных Т-клеток и появления аутореактивных Т-клеток. Эндокринопатии, наиболее часто встречающиеся при IPEX, — СД-1 с ранним дебютом и аутоиммунный тиреоидит. Любой случай диагностики СД-1 <6-9 мес обязательно должен побуждать к размышлению о наличии моногенного АПС/генетической причины дисфункции В-клеток. Пациенты с IPEX часто страдают аутоиммунной энтеропатией и экзематозным дерматитом; у них также м.б. др. аутоиммунные заболевания (напр., печени, почек, цитопении) и аллергические расстройства (напр., пищевая аллергия, эозинофиллия).
Лечение IPEX: иммуномодуляция с помощью иммунодепрессантов (напр., ГКС, такролимус), новых ЛП (напр., абатацепт), трансплантации стволовых клеток.
г) Другие моногенные синдромы иммунной дисрегуляции. Др. расстройства, вызванные нарушением периферической толерантности и возникновением аутоиммунных нарушений, часто сопровождаются иммунодефицитом. Эти нарушения, обусловленные мутациями с потерей функции в CD25 (IL2RA), LRBA, CTLA4, STAT5b и мутациями с приобретением функции в STAT1, патофизиологически сходны с IPEX. В целом пациенты с этими IPEX-подобными расстройствами, как правило, подвержены высокому риску развития СД-1 и аутоиммунного тиреоидита (см. табл. 1).
Они страдают разл. неэндокринными заболеваниями, особенно аутоиммунными и иммунодефицитными, с поражением кожи, легких и ЖКТ. STAT5b участвует в преобразовании сигнала IL2/STAT5, необходимого для активации сигнального пути гормона роста, а также может влиять на секрецию пролактина; поэтому пациенты с дефектами STAT5b могут иметь неаутоиммунную резистентность к гормону роста и гиперпролактинемию в дополнение к иммунной дисрегуляции, гипергаммаглобулинемии и множественным аутоиммунным расстройствам. Мутации, ведущие к появлению новой аномальной функции STAT1 подавляют нормальную продукцию цитокинов Thl7, что приводит к хроническому кандидозу слизистых оболочек.
Эти пациенты имеют высокий риск развития инфекционных заболеваний, плоскоклеточного рака, энтеропатии и артериальных аневризм. Пациенты с дефектами CD25 подвержены повышенному риску инфицирования, что обусловлено ролью сигнального пути IL-2 в формировании Th17-иммунных ответов.
д) Полигенный аутоиммунный полигландулярный синдром второго типа. АПС второго типа (АПС-2) — это клинический синдром, обусловленный наличием >2 специфических эндокринопатий: первичной недостаточности надпочечников аутоиммунного генеза (болезнь Аддисона), аутоиммунного заболевания ЩЖ (тиреоидит Хасимото/болезнь Грейвса) и/или СД-1. В некоторых классификациях АПС-2 подразделяют на подтипы в зависимости от вовлечения в патологический процесс определенных эндокринных желез (напр., подтипы 2, 3 и 4 включают поражения надпочечников/ЩЖ/ни одной из желез) либо в зависимости от др. имеющихся аутоиммунных проявлений (напр., подтипы ЗА, ЗВ и ЗС включают сопутствующие эндокринные, желудочно-кишечные и системные аутоиммунные нарушения). Однако, поскольку четкого патофизиол. различия между этими подтипами нет, мы рассматриваем их в совокупности как АПС-2.
Тем не менее при описании характеристик АПС-2 важно принимать во внимание некоторую степень совпадения между пациентами с клиническими проявлениями АПС-2 и пациентами с единичной эндокринопатией, сочетающейся с др. признаками аутоиммунной активности (напр., наличием аутоАТл), что в дальнейшем м.б. также классифицировано как АПС-2.
В отличие от моногенных форм АПС, которые обычно манифестируют в раннем детстве и имеют менделевский тип наследования, АПС-2 обычно проявляется во втором десятилетии жизни у пациентов с аутоиммунной эндокринопатией в личном анамнезе и семейной историей аутоиммунных заболеваний. АПС-2 наиболее распространен среди женщин среднего возраста (распространенность ок. 1:20 000). Могут наблюдаться первичная недостаточность гонад, витилиго, алопеция и хронический атрофический гастрит (с/без пернициозной анемии), однако аутоиммунный гипопаратиреоз и кандидоз нетипичны для АПС-2.
Хотя болезнь Аддисона встречается редко (распространенность ок. 1:10 000), пациенты с этим заболеванием подвергаются высокому риску развития др. эндокринных аутоиммунных расстройств, входящих в АПС-2; при этом сведения о наличии др. аутоиммунных болезней (в субклинической/клинической форме) опубликованы в 2/3 всех сообщений. Ок. половины всех пациентов с болезнью Аддисона имеют в качестве сопутствующего аутоиммунное заболевание ЩЖ (синдром Шмидта (Schmidt)), а 15% — СД-1 (синдром Карпентера (Carpenter)). Др. аутоиммунные проявления у 5-10% пациентов включают болезнь Грейвса, недостаточность яичников, алопецию, витилиго, пернициозную анемию, наличие аутоАТл, характерных для целиакии. АПС-2 реже развивается у пациентов СД-1, чем у пациентов с болезнью Аддисона; тем не менее у них часто наблюдаются др. аутоиммунные болезни.
Среди этих пациентов аутоиммунные заболевания ЩЖ/органов ЖКТ (каждое из этих состояний наблюдается у 20% пациентов) встречаются гораздо чаще, чем сопутствующая патология надпочечников (<1%). Тироксин и кортизол оказывают влияние на чувствительность к инсулину, метаболизм и аппетит, поэтому необъяснимая гипогли-кемия/ухудшение показателей гликемии м.б. первыми клиническими признаками АПС-2 у пациентов с уже имеющимся СД-1. Необъяснимая гипогликемия также может свидетельствовать о манифестации целиакии. В действительности целиакия зачастую предшествует развитию аутоиммунных эндокринопатий, включая СД-1, гипотиреоз и болезнь Аддисона.
АПС-2 относительно редко развивается у лиц с изолированным аутоиммунным заболеванием ЩЖ. Тем не менее врач должен учитывать вероятность недостаточности надпочечников у пациента с признаками, указывающими на АПС-2, до начала лечения первичного аутоиммунного гипотиреоза, т.к. заместительная терапия тиреоидными гормонами в этой ситуации может спровоцировать развитие надпочечникового криза. Как и в случае АПС-1, при обнаружении аутоАТл к определенным тканям м.б. инициировано проведение функционального скрининга еще до возникновения явных клинических проявлений заболевания (см. табл. 2); однако прогностическая ценность наличия этих аутоАТл для развития клинического заболевания весьма вариабельна.
Аномальные ответы Т-клеток, вероятно, играют определенную роль в патогенезе множественного поражения эндокринных желез, наблюдающегося при АПС-2. Вероятность развития аутоиммунных реакций, направленных против надпочечников, ЩЖ и островковых клеток, судя по всему, является общим для определенных гаплотипов HLA и др. генетических локусов, связанных с иммунитетом, хотя степень этого риска существенно варьирует для каждой эндокринопатии. У пациентов с АПС-2 наблюдается более высокая распространенность аллелей HLA-D3 и HLA-D4, что, видимо, увеличивает риск развития этого заболевания.
Определенные аллели основных генов А и В, связанных с МНС класса I (MICA и MICB), также участвуют в развитии АПС-2. Полиморфизмы др. генов (напр., PTPN22, CTLA4) способствуют развитию отдельных аутоиммунных эндокринопатий, составляющих АПС-2, однако вклад этих генов в патогенез самого АПС-2 остается неопределенным. Вероятно, существуют факторы окружающей среды, провоцирующие и способствующие развитию аутоиммунных заболеваний у генетически детерминированных лиц. Кроме того, многие из факторов риска, вызывающих эндокринные и неэндокринные аутоиммунные заболевания, пересекаются (см. отдельные статьи на сайте, посвященные этим заболеваниям, для более подробного обсуждения факторов риска - просим Вас пользоваться формой поиска по сайту выше).
е) Хромосомные аномалии, вызывающие развитие аутоиммунных полигландулярных синдромов. Многие генетические синдромы, обусловленные де-лециями, дупликациями и числовыми аномалиями хромосом, способствуют повышению риска развития аутоиммунной патологии, особенно аутоиммунных эндокринных болезней, поражающих ЩЖ и β-клетки ПЖЖ (см. табл. 1). К ним относятся синдром Тернера, синдром Кляйнфельтера, синдром ДиДжорджи (делеция 22q11.2) и трисомия 21 хромосомы. Мужчины с синдромом Кляйнфельтера и женщины с синдромом Тернера имеют повышенный риск развития аутоиммунных заболеваний многих систем, включая аутоиммунные эндокринные расстройства. Механизм формирования аутоиммунных реакций при трисомии 21-й хромосомы остается неясным, хотя были описаны различия в экспрессии гена AIRE, чувствительности к HLA, пролиферации аутоАТл.
Дисплазия тимуса является типичной особенностью синдрома ДиДжорджи, и возникающие в результате этого нарушения иммунной регуляции могут играть определенную роль в повышении риска развития аутоиммунных заболеваний при этом расстройстве. Пациенты с генетическими синдромами и хромосомными аномалиями могут иметь и неаутоиммунные эндокринопатии, такие как нарушение роста, первичную недостаточность гонад и первичный гипопаратиреоз.
Митохондриальные болезни редко вызывают развитие синдромов аутоиммунной эндокринопатии/полиэндокри-нопатии. Синдром Кэрнса-Сэйра (Kearns, Sayre) (прогрессирующая наружная офтальмоплегия, пигментный ретинит, нарушения сердечной проводимости) может сопровождаться аутоиммунными заболеваниями ЩЖ, надпочечников и СД. Др. мутации митохондриальных генов, включая MELAS (митохондриальная энцефаломиопатия, лактоацидоз, инсультоподобные эпизоды), сопровождаются отдельными эндокринными расстройствами, такими как недостаточность гонад, гипогонадизм, гипопаратиреоз, болезнь Аддисона и СД-1.
ж) Негенетические аутоиммунные причины полигландулярного эндокринного синдрома. Быстро прогрессирующее ожирение с гипоталамической дисфункцией, гиповентиляцией и вегетативной дисрегуляцией (ROHHAD*) — это редкий синдром, развивающийся у детей и диагностируемый по основным клиническим признакам. ROHHAD-синдром обычно дебютирует быстрым набором МТ у ранее здорового ребенка (возраст манифестации заболевания 1-9 лет).
P.S. * ROHHAD (Rapid-Onset Obesity with Hypothalamic Dysfunction, Hypoventilation, And Autonomic Dysregulation) — быстро прогрессирующее ожирение с гипоталамической дисфункцией, гиповентиляцией и вегетативной дисрегуляцией.
Со временем клиническая картина развивается, включая признаки вегетативной дисфункции (напр., офтальмологические симптомы, нарушение моторики ЖКТ, нарушение терморегуляции, брадикардию), центральную гиповентиляцию и разл. гипоталамические расстройства, такие как центральный гипотиреоз, дефицит гормона роста, гиперпролактинемия, гипонатриемия. Гипотеза о том, что ROHHAD-синдром имеет аутоиммунную паранеопластическую этиологию, подтверждается наличием маркеров иммуноопосредованного повреждения, его реакцией на иммуносупрессивную терапию у некоторых пациентов, а также связью этого синдрома с нейроэндокринными опухолями. Однако такие опухоли присутствуют только у 40% пациентов, причем удаление новообразования не может повлиять на процесс прогрессирования болезни. На сегодняшний день генетическая причина ROHHAD-синдрома не найдена.
Новейшие иммуномодулирующие биологические соединения все шире используются при лечении ЗНО и иммунных нарушений. Применение противоопухолевых ЛП, подавляющих контрольные точки иммунного ответа, такие как CLTA4, PD1 и PDL1, приводит к острой манифестации множественных аутоиммунных эндокринопатий, включающих гипофизит с гипопитуитаризмом, СД-1, первичную недостаточность надпочечников и тиреоидит. АТл к CD52, используемые при лечении рассеянного склероза, обусловливают развитие болезни Грейвса и др. АТд-опосредованных аутоиммунных заболеваний (напр., ИТП). Предсуществующий аутоиммунный процесс может являться фактором риска развития аутоиммунного заболевания после воздействия широкого спектра иммуномодулирующих ЛП.