При этом состоянии обнаруживается генотип XY, однако НПО либо не полностью вирилизированы, выглядят неоднозначно (атипичны), либо полностью развиты по женскому типу. В случае, когда гонады обнаруживаются, они обычно содержат элементы яичек; степень их развития колеблется в пределах от рудиментарного до нормального.
Поскольку нормальный процесс вирилизации у плода очень сложен, факт того, что существует множество разновидностей и причин НФП 46,XY, не вызывает удивления. Причина развития НФП 46,XY не определяется в 50% и < случаев.
а) Дефекты дифференцировки яичек. Первый этап мужской по мужскому типу — превращение бипотенциальной гонады в яичко. Если у плода с кариотипом XY имеется делеция короткого плеча Y-хромосомы или гена SRY, дифференцировка по мужскому типу не происходит. Фенотип — женский; мюллеровы протоки хорошо развиты из-за отсутствия АМГ, а половые железы представлены недифференцированными тяжами.
Напротив, у нормально развитых мужчин обнаруживались даже крайне обширные делеции длинного плеча Y-хромосомы (Yq-), у большинства таких мужчин отмечаются азооспермия и низкий рост.
Это указывает на то, что на длинном плече У-хромосомы в норме располагаются гены, предотвращающие эти проявления. В отношении многих синдромов, при которых яички не дифференцируются, при кариотипировании У-хромосомы морфологически нормальны.
- Синдром Дениса-Драша: основными клиническими проявлениями данного синдрома являются сочетание нефропатии с неоднозначными половыми органами и двусторонней WT1. Большинство зарегистрированных случаев обладали кариотипом 46,ХУ. Часто присутствуют мюллеровы протоки, что указывает на общую недостаточность функции яичек плода. Пациенты с кариотипом 46,XX имеют нормальные НПО. Протеинурия, возникающая в младенчестве, прогрессирует до развития нефротического синдрома и терминальной стадии почечной недостаточности к 3 годам жизни, причем наиболее систематически выявляемым результатом гистопатологического исследования является очаговый или диффузный мезангиальный склероз.
WT1 обычно развивается у детей <2 лет и часто бывает двусторонней. Также сообщалось об обнаружении гонадобластом.
Было обнаружено несколько мутаций гена WT1, расположенного на хромосоме 11p13. WT1 функционирует как ген-супрессор опухоли и фактор транскрипции и экспрессируется в генитальном гребне и фетальных гонадах. Почти все зарегистрированные мутации происходили вблизи или в пределах области, кодирующей цинковый палец. В одном сообщении была обнаружена мутация домена цинкового пальца в аллелях WT1 у пациента без пороков развития МВП, что позволяет предположить, что некоторые спорадические случаи WT1 могут иметь мутацию WT1.
- Синдром Фрейзера: при синдроме Фрейзера, состоянии, характеризующемся неспецифическими очаговым и сегментарным гломерулосклерозом, XY дисгенезией гонад и частым развитием гонадобластомы, но без WT1, были описаны разл. мутации гена WT1, конституциональные гетерозиготные мутации в интроне 9.
- Синдром WAGR: данная аббревиатура обозначает синдром «генных последовательностей», состоящий из WT1, аниридии, пороков развития МПС и задержки интеллектуального развития. У таких детей присутствует делеция одной копии хромосомы 11p13, которую можно увидеть при кариотипировании. В области делеции располагаются ген аниридии (РАХ6) и ген-супрессор WT1. Генитальные аномалии, начиная от крипторхизма и заканчивая тяжелой недостаточностью вирилизации, наблюдаются только у пациентов с кариотипом 46,ХY.
В дисгенетических половых железах развиваются гонадобластомы. WT1 обычно возникает к двум годам жизни. В некоторых случаях также наблюдалось необъяснимое ожирение, в связи с чем возник вопрос о наличии в этой области хромосомы 11 гена, ассоциированного с ожирением и названии синдрома WAGRO.
2. Кампомелическая дисплазия. Эта форма скелетной дисплазии с короткими конечностями характеризуется искривлением бедренной и большеберцовой костей кпереди, небольшими гипопластическими лопатками, небольшой грудной полостью и 11 парами ребер и с пороками развития др. органов. Обычно она приводит к летальному исходу в раннем младенческом возрасте. У ~75% из зарегистрированных пациентов с кариотипом 46XY фенотип полностью женский; наружные и внутренние половые органы — женские. У некоторых пациентов с кариотипом 46,XY отмечаются неоднозначные половые органы. Половые железы, по-видимому, представлены яичниками, но гистологически могут содержать элементы как яичников, так и яичек.
Ген, ответственный за это состояние, — SOX9 [связанный с SRY ген HMG-бокса (группы белков с высокой подвижностью)], он находится на 17q24-q25. Этот ген структурно связан с SRY, а также непосредственно регулирует развитие гена коллагена типа II (COL2A1). Одни и те же мутации могут приводить к различным фенотипам половых желез. У пациента с этим заболеванием зарегистрировано развитие гонадобластомы. Тип наследования — АуД.
3. Дефекты SF-1 (также известного как Ad4BP или NR5A1) и нарушение полового развития 46,XY. У пациентов с мутациями гена SF-1 были описаны надпочечниковая недостаточность и 46,XY дисгенезия гонад. В некоторых случаях, если мать этих пациентов тоже имеет мутацию SF-1, у нее отмечается преждевременная недостаточность яичников, так же как и в описаниях матерей младенцев с НПР 46,XX. НПР 46,XY, связанное с SF-1, также может развиваться без проявлений надпочечниковой недостаточности и напоминать синдром частичной нечувствительности к андрогенам.
Инверсия пола при кариотипе 46,ХУ также была описана у пациентов с делециями частей аутосомных локусов на хромосомах 2q, 9р и 10q.
4. Чистая дисгенезия гонад XY (синдром Свайера). Название «чистая» отличает это состояние от форм дисгенезии гонад, имеющих хромосомное происхождение и связанных с соматическими аномалиями. Пациенты, страдающие этим заболеванием, имеют нормальный рост и женский фенотип, в т.ч. влагалище, матку и фаллопиевы трубы, но в пубертатном возрасте не происходит развитие МЖ и не наступает менархе. Не отмечается никаких др. фенотипических признаков, связанных с 45,X (синдром Тернера). Клиническая картина у пациентов развивается в период полового созревания и характеризуется гипергонадотропной первичной аменореей.
Семейные случаи позволяют предположить сцепленный с Х-хромосомой или ограниченный полом АуД-тип наследования.
У большинства обследованных пациентов отмечались мутации гена SRY. Половые железы представлены почти полностью недифференцированными тяжами, несмотря на наличие цитогенетически нормальной Y-хромосомы. Примитивная гонада не может осуществлять никаких функций яичек, в т.ч. подавление развития мюллеровых (парамезонефральных) протоков. В гонаде могут присутствовать хилюсные кл., способные продуцировать некоторое количество андрогенов. Соответственно, в период полового созревания может произойти некоторая вирилизация, напр. увеличение клитора.
Половые железы, представленные тяжом, могут подвергаться опухолевым изменениям, как, напр., развитие гонадобластом и дисгермином, и должны удаляться сразу же после постановки диагноза, независимо от возраста пациента.
Чистая дисгенезия гонад также встречается у людей с кариотипом XX.
5. Синдром XY агенезии гонад (синдром эмбриональной регрессии яичек). При этом редком синдроме НПО выглядят слегка неоднозначно, но более близки к женским. Присутствуют гипоплазия половых губ, некоторая степень сращения половых губ, маленький клитороподобный пенис и уретральное отверстие, открывающееся в промежность. Не обнаруживаются матка, ткань половых желез и зачастую влагалище. В пубертатном возрасте полового развития не происходит, а уровень гонадотропных гормонов повышен. Большинство детей воспитываются как женщины.
У нескольких пациентов с ХY агенезией гонад, у которых половые железы не были обнаружены при эксплоративной операции, стимуляция ХГЧ приводила к значительному повышению уровня тестостерона, что указывало на присутствие где-то в организме функционирующих кл. Лейдига. Известны случаи этого расстройства у братьев и сестер.
Предполагается, что ткань яичек была активна в течение жизни плода на протяжении времени, достаточного для ингибирования АМГ развития мюллеровых протоков, но недостаточного для выработки тестостерона, приводящей к вирилизации. У одного пациента не было обнаружено делеции Y-хромосомы при исследовании с помощью Y-специфических ДНК-зондов. Дегенерация яичек, вероятно, происходит между 8-й и 12-й неделями в/утробной жизни плода. Регрессия яичка в период до 8-й недели гестации приводит к синдрому Свайера, между 14-й и 20-й неделями гестации — к синдрому рудиментарных яичек, а после 20-й недели — к анорхизму.
При двустороннем анорхизме, иногда называемом синдромом «исчезающих» яичек, яички отсутствуют, но наблюдается полностью мужской фенотип; предполагается, что ткань с функцией яичек была активна у плода в критический период половой дифференцировки, но некоторое время спустя она была повреждена. Двусторонний анорхизм у однояйцевых близнецов и односторонний анорхизм у однояйцевых близнецов и у сиблингов указывают на генетическую предрасположенность.
Сосуществование анорхизма и синдрома агенезии гонад у детей одних родителей свидетельствует о взаимосвязи между этими расстройствами. О дефектах SRY у пациентов с анорхизмом еще не сообщалось.
Ретроспективный обзор урологических эксплоративных операций 691 яичек выявил отсутствие яичек в 21% случаев. Из них в 73% обнаруживались слепо заканчивающиеся тяжевидные структуры, при этом предполагаемым местом исчезновения яичек были паховый канал (59%), БП (21%), поверхностное паховое кольцо (18%) и мошонка (2%). Было высказано предположение, что обнаружение при лапароскопии тяжевидных структур должно побудить к эксплоративной операции паховой области, поскольку у четырех из этих детей была обнаружена жизнеспособная ткань яичек. Никаких данных о гормонах (стимуляционная проба с ХГЧ, уровень АМГ) не поступало.
б) Недостаточность выработки гормонов яичка. Было определено несколько генетических дефектов в ферментативном синтезе тестостерона яичками плода, также был описан дефект в дифференцировке кл. Лейдига. Эти дефекты приводят к неадекватной маскулинизации у мужчин с кариотипом 46,XY. Поскольку уровень тестостерона до начала полового созревания обычно низок, для оценки способности яичек к синтезу тестостерона у детей может потребоваться стимуляционная проба с ХГЧ.
1. Апплазия клеток Лейдига. Пациенты с аплазией или гипоплазией кл. Лейдига обычно имеют женский фенотип, но может наблюдаться и умеренная вирилизация. Обнаруживаются яички, придатки яичка и семявыносящие протоки; матка и фаллопиевы трубы отсутствуют из-за нормальной выработки АМГ. В период полового созревания не происходит развития МЖ, однако развитие лобковых волос м.б. нормальным из-за выработки надпочечниковых андрогенов. Уровень тестостерона в плазме крови низкий и не реагирует на ХГЧ; уровень ЛГ повышен. Кл. Лейдига в яичках отсутствуют или наблюдается их значительный дефицит. Данный дефект м.б. связан с отсутствием функциональных рецепторов к ЛГ.
У детей необходимо проведение стимуляционной пробы с ХГЧ, чтобы отличить это состояние от синдромов нечувствительности к андрогенам. Тип наследования — ограниченный мужским полом АуР. Рецептор человеческого ЛГ/ХГЧ является членом суперсемейства рецепторов, сопряженных с G-белком, которые содержат семь трансмембранных доменов. Было описано несколько инактивирующих мутаций рецептора к ЛГ/ХГЧ у мужчин с гипогонадизмом, подозрением на гипоплазию или аплазию кл. Лейдига.
У одного мужчины с гипогонадизмом отмечались высокий уровень сывороточного ЛГ и низкий уровень ФСГ по причине мутации в гене β-субъединицы ФСГ (см. табл. ниже).
2. Липоидная гиперплазия надпочечников. Это самая тяжелая форма врожденной гиперплазии надпочечников, она получила свое название из-за обнаружения увеличенных надпочечников в результате накопления холестерина и эфиров холестерина. Процессом, лимитирующим скорость реакции в стероидогенезе, является транспорт свободного холестерина через цитозоль к внутренней митохондриальной мембране, где действует фермент расщепления боковой цепи Р450 (P450scc; CYP11A1). Транспорт холестерина в митохондрии опосредуется стероидогенным острым регуляторным белком (StAR). StAR — это белок с Mr 30 кД, необходимый для стероидогенеза и кодируемый геном, расположенным на хромосоме 8р11.2.
Митохондриальное содержание StAR увеличивается во временном промежутке между первым и пятым часом после стимуляции адренокортикотропным гормоном, спустя долгое время после окончания резкого усиления стероидогенеза, индуцированного АКТГ. Это привело к предположению, что экстрамитохондриальный StAR также может принимать участие в быстром ответе на АКТГ. У большинства пациентов с липоидной врожденной гиперплазией коры надпочечников имеются мутации в гене, кодирующем StAR, а у некоторых имеются мутации в CYP11A1.
Уровни всех стероидных гормонов в сыворотке крови низкие или не определяются, в то время как уровни АКТГ и ренина в плазме крови значительно повышены. У лиц как с женским, так и с мужским генотипом фенотип женский. Лица с мужским генотипом не имеют структур, развивающихся из мюллеровых протоков, потому что яички могут производить нормальный АМГ, но не вырабатывают стероидные гормоны. У таких детей в раннем детстве наблюдается острый адреналовый криз и потеря соли. Большинство пациентов обладают генотипом 46,XY. У некоторых пациенток в период полового созревания наблюдается стероидогенез в яичниках.
Примером регуляторной роли стероидогенеза, независимого от StAR, является клинический случай 4-месячных близнецов с кариотипом 46,XX с липоидной гиперплазией надпочечников. Одна умерла в 15 мес из-за осложнений со стороны ССС, связанных с коарктацией аорты. В надпочечниках имелись характерные липидные отложения. У выжившей девочки-близнеца наступило спонтанное половое созревание с феминизацией в 11,5 года и менархе в 13,8 года. При повторном обследовании в возрасте 15 лет у нее была обнаружена гомозиготная инактивирующая мутация со сдвигом рамки считывания в гене StAR.
Это и тот факт, что она выжила в младенчестве до 4-месячного возраста без заместительной терапии с определяемым уровнем альдостерона в сыворотке крови, подтверждают гипотезу о том, что стероидогенез, независимый от StAR, мог происходить до тех пор, пока не накопится количество в/кл. липидов, достаточное для угнетения стероидогенной активности. Описаны дефекты с частичной инактивацией гена у лишь частично вирилизированных мужчин и при отсроченном начале потери соли. Дефекты, приводящие к полной инактивации CYP11A1, могут быть несовместимы с жизнью, поскольку только этот фермент может преобразовывать холестерин в прегненолон, затем превращающийся в прогестерон, гормон, необходимый для поддержания нормальной беременности у млекопитающих.
У 4-летнего ребенка с инверсией пола при кариотипе 46,XY и формой липоидной гиперплазии надпочечников с отсроченным началом была описана гетерозиготная мутация в CYP11A1.
На 6-7-й неделе гестации, когда синтез прогестерона в желтом теле матери прекращается, плацента, не экспрессирующая StAR, синтезирует прогестерон путем StAR-независимого стероидогенеза с использованием ферментной системы CYP11A1.
3. Недостаточность 3β-гидроксистероид-дегидрогеназы. У мужчин с данной формой врожденной гиперплазии надпочечников отмечаются гипоспадии различной степени с или без расщепления мошонки и крипторхизма и, редко, полностью женский фенотип. У младенцев с таким заболеванием обычно вскоре после рождения развиваются симптомы солевых потерь. Сообщалось о дефектах с неполной недостаточностью фермента, иногда наблюдаемых у мужчин с преждевременным пубархе, а также о неклассических формах с поздним началом. Такие дети имеют точечные мутации гена 3β-гидроксистероидного фермента типа II, приводящие к нарушению стероидогенеза в надпочечниках и половых железах; недостаточность в надпочечниках и половых железах может проявляться неравномерно.
Нормальные изменения, характерные для полового созревания, отмечающиеся у некоторых мужчин, можно объяснить нормальным содержанием 3β-гидроксистероид-дегидро-геназы типа I, которая присутствует во многих периферических тканях. Часто встречается бесплодие. Корреляции между степенью потери соли и выраженностью фенотипических аномалий не существует.
4. Недостаточность 17-гидроксилазы/17,20-лиазы. Один фермент (CYP17A1), кодируемый одним геном, расположенным на хромосоме 10q24.3, обладает как 17-гидроксилазной, так и 17,20-лиазной активностью в тканях надпочечников и половых желез. Зарегистрировано множество различных генетических мутаций. Лица с мужским генотипом обычно обладают полностью женским фенотипом, или, что реже, у них отмечается недостаточная вирилизация различной степени от сращения половых губ до промежностной гипоспадии и крипторхизма. Половое развитие в пубертатном периоде не происходит у обоих генетических полов.
При классической форме заболевания наблюдается ↓ синтеза кортизола надпочечниками и половых стероидов надпочечниками и гонадами. Уровни предшественника стероидных гормонов, обладающего минералокортикоидной активностью, дезоксикортикостероном, а также кортикостероном, заметно повышены, что приводит к гипертензии и гипокалиемии, характерным для этой формы 46,XY. Несмотря на то, что уровень кортизола низок, повышенные уровни АКТГ и кортикостерона предотвращают клинические проявления недостаточности кортизола. Ренин-альдостероновая ось подавляется в результате выраженного минералокортикоидного эффекта высокого уровня дезоксикортикостерона.
В период полового созревания вирилизация не происходит; уровень тестостерона низок, а уровень гонадотропных гормонов повышен.
Поскольку продукция АМГ плодом в норме, рудиментарные остатки мюллерова протока не определяются. У пациентов с женским фенотипом и кариотипом XY показана гонадэктомия и заместительная терапия гидрокортизоном и половыми стероидами.
Дефект обладает АуР-типом наследования. Женщины с кариотипом XX и данным заболеванием обычно не выявляются до ранней стадии взросления, когда у них не отмечается изменений, типичных для полового развития, и не обнаруживается АГ и гипокалиемия. Такое заболевание следует заподозрить у пациенток с первичной аменореей и АГ, имеющих хромосомный набор 46,XX или 46,XY.
Некоторые пациенты, у которых изначально была описана изолированная недостаточность 17,20-лиазы, как было показано впоследствии, имели дефект синтеза ДГТ из-за недостаточности ферментов в альтернативном пути синтеза ДГТ (данное состояние более подробно описано ниже).
5. Недостаточность 17-кетостероидредуктазы. Этот фермент, также называемый 17β-гидроксистероид-дегидрогеназой, катализирует заключительную стадию биосинтеза тестостерона. Он необходим для того, чтобы преобразовать андростендион в тестостерон, дегидроэпиандростерон в андростендиол и эстрон в эстрадиол. Недостаточность 17-кетостероидредуктазы в яичке плода приводит к тому, что мужской плод имеет полностью или почти полностью женский фенотип. Мюллеровы протоки отсутствуют, а влагалище неглубокое. Диагноз ставится на основании соотношения андростендиона и тестостерона. У детей препубертатного возраста для постановки диагноза может потребоваться стимуляционная проба с ХГЧ.
Для данного дефекта характерен АуР-тип наследования. Идентифицировано по крайней мере четыре различных типа 17(3-гидроксистероид-дегидрогеназы, каждый из которых кодируется отдельным геном, расположенным на разных хромосомах. Тип III — фермент, отвечающий за выработку тестостерона в яичках. Этот дефект чаще встречается у арабского населения Газы с более высокой степенью родственных браков, чем у др. групп населения. Ген, кодирующий это расстройство, находится на 9q22 и экспрессируется только в яичках, где он преобразует андростендион в тестостерон.
Большинству пациенток диагноз ставится в период полового созревания из-за вирилизации и отсутствия менструаций. Уровень тестостерона в период полового созревания м.б. почти нормальным, предположительно в результате периферического превращения андростендиона в тестостерон; в этот период некоторые пациенты могут спонтанно принять на себя мужскую гендерную роль.
17β-гидроксистероид-дегидрогеназа типа I, кодируемая геном, расположенным на хромосоме 17q21, преобразует эстрон в эстрадиол и обнаруживается в плаценте, яичниках, яичках, печени, предстательной железе, жировой ткани и эндометрии. Тип II, ген которого находится на хромосоме 16q24, катализирует обратную реакцию для типов I и III (превращение тестостерона в андростендион и эстрона в эстрадиол соответственно). Тип IV по своему действию аналогичен типу II. Форма недостаточности 17-кетостероидредуктазы с поздним началом проявляется в форме гинекомастии у молодых взрослых мужчин.
6. Синдром персистенции мюллеровых протоков. При этом заболевании наблюдается персистенция производных мюллеровых (парамезонефральных) протоков у полностью вирилизованных мужчин. Сообщалось о случаях заболевания у сиблингов и однояйцевых близнецов. Крипторхизм наблюдается у 80% пораженных мужчин; когда во время операции по данному поводу или по поводу паховой грыжи обнаруживаются маточная труба и матка, о заболевании становится известно. Степень развития мюллеровых протоков различна и м.б. асимметричной.
Функция яичек в большинстве случаев нормальная, однако есть сообщения о дегенерации яичек. У некоторых мужчин с таким заболеванием после наступления полового созревания развиваются опухоли яичек. В исследовании, в котором приняло участие 38 семей, 16 семей имели дефекты в гене АМГ, расположенном на коротком плече хромосомы 19. У пациентов с такой мутацией отмечался низкий уровень АМГ.
В 16 семьях, у членов которых был высокий уровень АМГ, дефект был обнаружен в гене рецептора к АМГ типа II, причем в 10 из 16 случаев отмечались идентичные делеции, размером 27 пар оснований, на экзоне 10 по крайней мере в одном аллеле.
Лечение заключается в удалении как можно большего количества структур, развивающихся из мюллеровых протоков, не повреждая яичко, придаток яичка или семявыносящий проток.
в) Нарушения действия андрогенов. Следующая группа заболеваний характеризуется нормальным фетальным синтезом тестостерона, а нарушение вирилизации является результатом наследуемых аномалий в действии андрогенов.
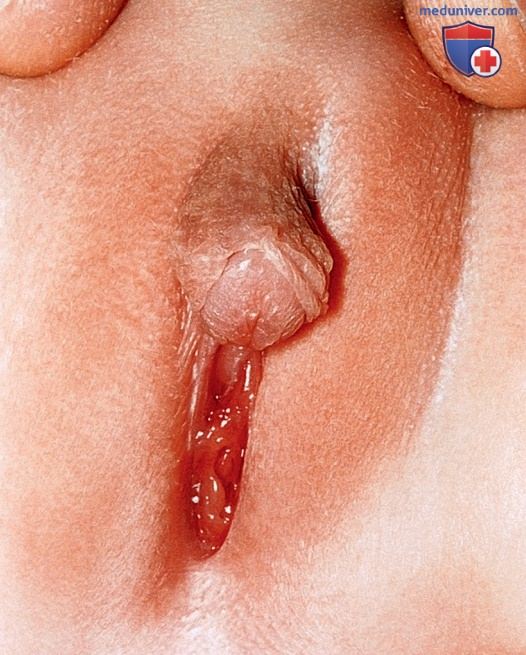
1. Недостаточность дигидротестостерона. Снижение продукции ДГТ в период в/утробного развития приводит к выраженной неоднозначности НПО у мужчин с этим заболеванием. Биосинтез и периферическое действие тестостерона в норме.
Фенотип, который наиболее часто встречается при этим состоянии, приводит к появлению у мужчин небольшого пениса, расщепленной мошонки, урогенитального синуса с промежностной гипоспадией и слепо оканчивающимся влагалищем (рис. ниже). Яички находятся в паховом канале или в губно-мошоночных складках и не имеют гистологических отклонений. Структуры, развивающиеся из мюллеровых протоков, не определяются. Структуры, развивающиеся из вольфовых протоков, — семявыносящий проток, придаток яичка и семенные пузырьки — присутствуют. Пол большинства пациентов с данным заболеванием был первоначально определен как женский.
Недостаточность 5α-редуктазы
В период полового созревания происходит вирилизация; пенис увеличивается, яички опускаются и нормально растут, и происходит сперматогенез. Гинекомастия отсутствует. Волосы бороды растут скудно, угревая сыпь отсутствует, предстательная железа маленькая, также не отмечается височного облысения. Вирилизация вольфова протока происходит под действием самого тестостерона, однако маскулинизация урогенитального синуса и НПО зависит от действия ДГТ в критический период маскулинизации плода. Рост волос на лице и рост простаты также зависят от ДГТ.
Рост, достигаемый ко взрослому возрасту, близок к росту отца и братьев. Отмечается значительная фенотипическая гетерогенность. Это привело к распределению таких пациентов на пять типов недостаточности стероидной 5α-редуктазы.
Во всем мире у пациентов было выявлено несколько разл. дефектов гена SRD5A2 (ген 5α-редуктазы типа 2, приводящий к дефициту 5α-редуктазы), расположенных на коротком плече хромосомы 2. Сообщения о семейных группах поступали из Доминиканской Республики, Турции, Папуа-Новой Гвинеи, Бразилии, Мексики и стран Ближнего Востока. Уверенной корреляции между генотипом и фенотипом не существует.
Данное расстройство наследуется по АуР-типу, но ограничивается мужским полом; нормальные гомозиготные женщины с нормальной репродуктивной способностью указывают на то, что у женщин ДГТ не играет клинически значимой роли при половой дифференцировке или в функции яичников в более позднем возрасте. Клинический диагноз должен быть поставлен как можно раньше в младенческом возрасте. Важно отличать данное состояние от синдрома частичной нечувствительности к андрогенам, потому что пациенты с синдромом частичной нечувствительности к андрогенам значительно менее чувствительны к андрогенам, чем пациенты с дефицитом 5α-редуктазы.
Биохимическая диагностика дефицита 5α-редуктазы основывается на обнаружении нормального уровня тестостерона в сыворотке крови, нормального или низкого уровня ДГТ при заметно повышенном базальном уровне тестостерона и, в частности, уровне тестостерона после стимуляции ХГЧ: коэффициентах ДГТ (>17) и высоком соотношении этиохоланолона в моче к андростерону. У детей с нечувствительностью к андрогенам, в отличие от детей с РПР, наблюдается нормальная 5α-редукция в печени и, следовательно, нормальное соотношение тетрагидрокортизола к 5α-тетрагидрокортизолу.
Важно отметить, что многие, но не все дети с дефицитом 5α-редуктазы, воспитанные в детстве как женщины, сменили пол на мужской в период полового созревания. По-видимому, воздействие тестостерона во в/утробном, неонатальном периодах и в период полового созревания вносит разный вклад в формирование мужской гендерной идентичности. Необходимо многое узнать о влиянии гормонов, таких как андрогены, а также о влиянии культурных, социальных, психологических, генетических и др. биологических факторов на гендерную идентичность и поведение. Младенцы с данным заболеванием должны воспитываться как мужчины в случаях, когда это практически целесообразно. Лечение младенцев мужского пола ДГТ приводит к увеличению пениса.
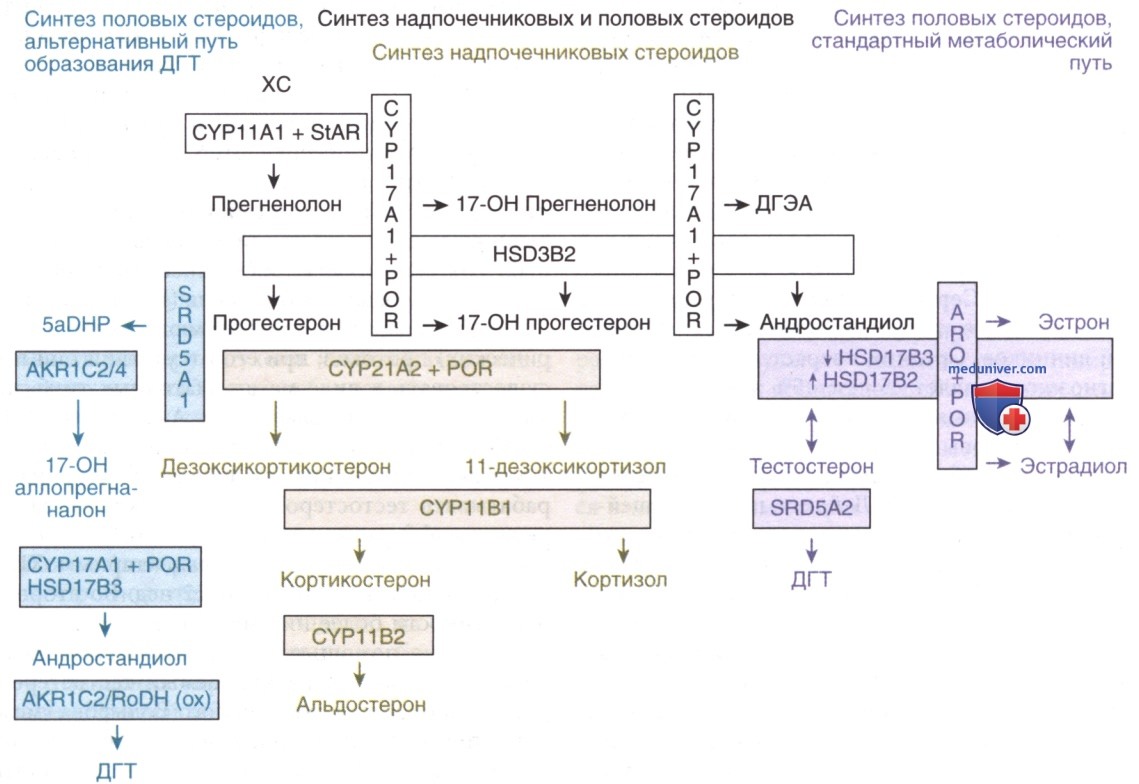
Др. причиной недостаточности ДГТ является блокада альтернативного пути синтеза ДГТ. Пациенты, у которых ранее считалось, что у них 46,XY DSD из-за изолированного дефицита 17,20-лиазы, впоследствии были охарактеризованы как имеющие мутации в гене AKR1C2 (3α-редуктаза 3-го типа) или обоих генах AKR1C2 и AKR1C4 (3α-редуктаза 4-го типа) (см. рис. ниже). Эти данные продемонстрировали, что для нормальной пренатальной вирилизации необходимо отсутствие повреждений как в классическом, так и в альтернативном путях синтеза ДГТ.
Названия генов и кодируемых ими ферментов путей стероидогенеза. CYP11А1: обеспечивает активность 20-гидроксилазы, 22-гидроксилазы и 20,22-лиазы, ответственных за расщепление боковой цепи холестерина. CYP17A1: обеспечивает активность 17α-гидроксилазы и 17,20-лиазы. 3βHSD2 (HSD3B2): обеспечивает активность 30-гидроксистероид-дегидрогеназы (тип 2) и D5D4-изомеразы. CYP21A2: активность 21-гидроксилазы. CYP11В1: активность 11β-гидроксилазы. CYP11В2: обеспечивает активность 18-гидроксилазы (CMOI) и 18-дегидрогеназы (CMOII). SRD5A1: активность 5α-редуктазы типа 1. SRD5A2: активность 5α-редуктазы типа 2. HSD17B2: активность 17β-гидроксистероид-дегидрогеназы типа 2. HSD17B3: активность 17β-гидроксистероид-дегидрогеназы типа 3. AKR1C2/4 (красный): активность 3α-редуктазы типов 1 и 3. AKR1C2/ToDH 9ох: активность Зα-редуктазы и 3-гидроксиэпимеразы. ARO — ароматаза; CMOI — кортикостеронметилоксидаза типа 1; CMOII — кортикостеронметилоксидаза типа 2; ДГЭА — дегидроэпиандростерон; ДГТ — дигидротестостерон; 5α-ДГП — 5α-дигидропрогестерон.
2. Синдромы нечувствительности к андрогенам. Синдромы нечувствительности к андрогенам — наиболее распространенные формы НФП у мужчин, встречающиеся с предполагаемой частотой 1/20 000 у лиц с мужским генотипом. Эта группа гетерогенных, сцепленных с Х-хромосомой рецессивных расстройств возникает в результате более чем 150 различных мутаций в гене андрогенного рецептора, расположенном на Xq11-12: точечных мутаций, приводящих к заменам аминокислот или образованию преждевременных стоп-кодонов, сдвигу рамки считывания и преждевременной терминации, делециям генов, а также к мутациям сайтов сплайсинга.
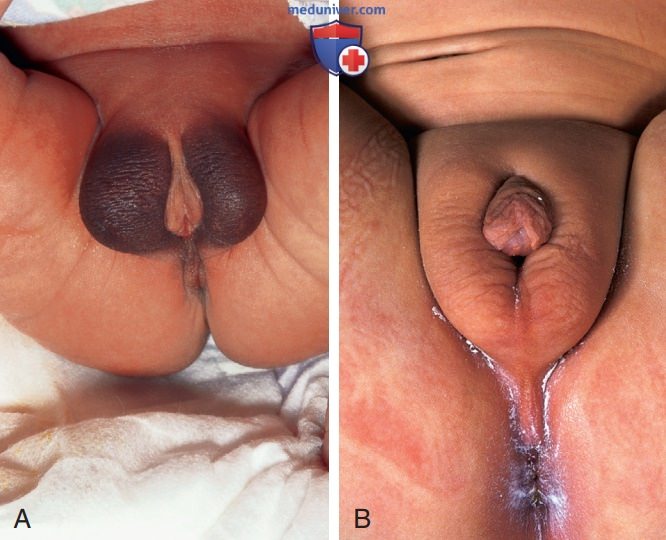
- Клинические проявления. Клинический диапазон пациентов с синдромами нечувствительности к андрогенам, каждый из которых имеет хромосомный набор 46,XY, варьируется от лиц с женским фенотипом (при полной нечувствительности к андрогенам) до мужчин с разл. формами неоднозначных гениталий и недостаточной вирилизации (частичная нечувствительность к андрогенам или клинические синдромы, такие как синдром Райфенстайна), а также до фенотипически нормальных мужчин с бесплодием. В дополнение к нормальному хромосомному набору 46,XY для всех таких детей характерно наличие яичек и нормальные или повышенные уровни тестостерона и ЛГ (рис. ниже).
А — частичная нечувствительность к андрогенам: опустившиеся яички в расщепленных губно-мошоночных складках; В — менее тяжелая форма частичной нечувствительности к андрогенам с тяжелой степенью гипоспадии и нарушением опущения яичек.
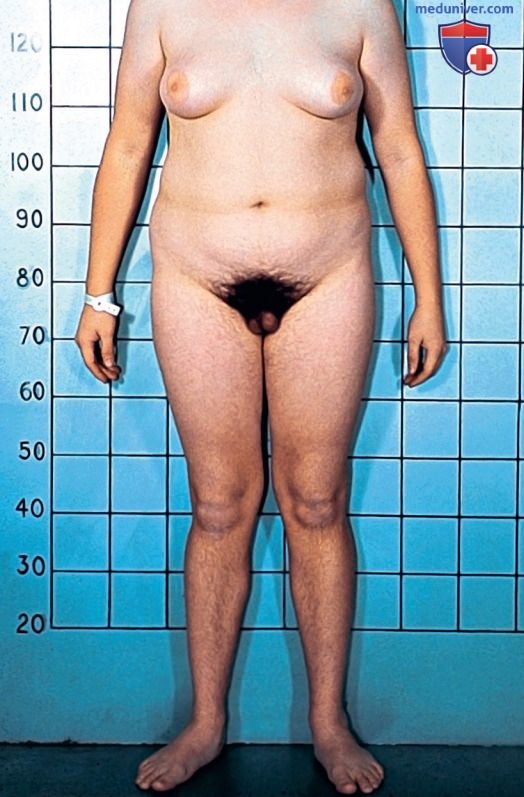
Синдром частичной нечувствительности к андрогенам в подростковом возрасте, воспитание по мужскому полу. Обратите внимание на гинекомастию, обусловленную периферическим превращением тестостерона в эстроген под действием ароматазы. Выраженный рост волос на лобке подразумевает лишь частичную нечувствительность.
При синдроме полной нечувствительности к андрогенам, крайнем проявлении нарушения вирилизации, дети, генетически являющиеся мужчинами, при рождении выглядят как девочки и неизменно воспитываются в соответствии с этим полом. НПО развиты по женскому типу. Влагалище представляет собой слепой мешок, а матка отсутствует в результате нормального синтеза яичками и действия АМГ. Примерно у трети пациентов обнаруживаются остатки маточных труб с одной или двух сторон. Яички обычно расположены в БП, но могут опускаться в паховый канал; они состоят в основном из семенных канальцев. В период полового созревания происходит нормальное развитие МЖ и внешне пациент напоминает женщину, однако менструации не начинаются, а половые волосы отсутствуют.
Несмотря на выраженную врожденную недостаточность эффекта андрогенов, рост взрослых соизмерим с ростом нормальных мужчин.
Яички пациентов с этим заболеванием, достигших зрелого возраста, вырабатывают тестостерон в нормальном для мужчины количестве, который преобразуется, соответственно, в нормальное количество ДГТ. Нарушение нормальной в/утробной дифференцировки по мужскому типу является отражением нарушенной реакции на андрогены в течение этого периода. Отсутствие андрогенных эффектов обусловлено поразительной устойчивостью к действию эндогенного или экзогенного тестостерона на кл. уровне.
До наступления полового созревания это расстройство часто выявляется у девочек, когда образования в паховом канале оказываются яичками или когда яичко неожиданно обнаруживается во время грыжесечения. 1-2% женщин с паховой грыжей страдают данным расстройством. Повышенный уровень ЛГ у младенцев должен наводить на мысль о данном диагнозе. Аменорея обычно является ведущим симптомом у детей старшего возраста и взрослых. У детей препубертатного возраста это состояние следует дифференцировать от др. заболеваний, проявляющихся недостаточной вирилизацией мужчин с кариотипом ХY, при которых наблюдается полная феминизация.
К данным состояниям относятся ХУ-дисгенезия гонад (синдром Свайера), истинный агонадизм, аплазия кл. Лейдига, в т.ч. дефекты рецепторов ЛГ, и дефицит 17-кетостероидредуктазы. Все эти состояния, в отличие от синдрома полной нечувствительности к андрогенам, характеризуются низким уровнем тестостерона как у новорожденных, так и у взрослых, а также отсутствием реакции на ХГЧ в препубертатном возрасте.
Несмотря на то, что пациенты с синдромом полной нечувствительности к андрогенам при рождении имеют однозначно женские НПО, у пациентов с синдромом частичной нечувствительности к андрогенам наблюдается широкий спектр фенотипических проявлений, начиная от промежностно-мошоночной гипоспадии, расщепленной мошонки и крипторхизма до крайне выраженной недостаточной вирилизации, проявляющейся в виде клиторомегалии и сращения половых губ. Некоторые формы синдрома частичной нечувствительности к андрогенам известны как специфические синдромы. У пациентов с синдромом Райфенстайна наблюдается неполная вирилизация, характеризующаяся гипогонадизмом, тяжелой степенью гипоспадии и гинекомастией (см. рис. выше). Синдромы Жильбер-Дрейфуса и Любса также относятся к категории синдрома частичной нечувствительности к андрогенам.
Во всех случаях были выявлены аномалии в гене андрогенного рецептора. В табл. 7 перечислены др. причины синдрома, напоминающего синдром частичной нечувствительности к андрогенам.
- Диагностика. В младенчестве поставить диагноз пациентам с синдромом частичной нечувствительности к андрогенам особенно трудно. Послеродовой всплеск тестостерона и ЛГ у пациентов с синдромом полной нечувствительности к андрогенам снижен, однако у пациентов с синдромом частичной нечувствительности к андрогенам этого не отмечается. У некоторых, особенно при достаточной степени вирилизации в младенчестве, диагноз не предполагается до наступления полового созревания, когда наблюдается неадекватная вирилизация с отсутствием волос на лице или изменения голоса и появлением гинекомастии.
Часто встречаются азооспермия и бесплодие. Все чаще дефекты андрогенных рецепторов выявляются у взрослых с маленьким пенисом и яичками, а также с бесплодием. Сообщалось о единичной аминокислотной замене в рецепторе андрогена в большой китайской семье, в которой некоторые члены, страдающие этим заболеванием, были фертильны, в то время как у др. наблюдалась гинекомастия и/или гипоспадии. При синдроме полной нечувствительности к андрогенам снижена продукция дермальными фибробластами гениталий инсулиноподобного фактора роста 2 и белка-2, связывающего инсулиноподобный фактор роста, но не белка-3, связывающего инсулиноподобный фактор роста, по сравнению с дермальными фибробластами гениталий в норме, что позволяет предположить возможную роль системы инсулиноподобного фактора роста в модуляции действия андрогенов.
- Лечение и прогноз. У пациентов с синдромом полной нечувствительности к андрогенам с однозначно женской сексуальной ориентацией яички следует удалять сразу же после их обнаружения. Лапароскопическое удаление гонад, содержащих генетический материал У-хромосомы, выполняется у пациентов с синдромом нечувствительности к андрогенам и у пациентов с дисгенезией гонад. У одной трети больных к 50-летнему возрасту развиваются ЗНО, как правило, семиномы. Семиномы развились у нескольких девочек-подростков. В период полового созревания показана заместительная терапия эстрогенами.
У женщин, яички которых не были удалены к возрасту начала полового созревания, развиваются нормальные МЖ. Продукция у них эстрадиола является результатом воздействия ароматазы на тестостерон, вырабатываемый яичками. Отсутствие андрогенного эффекта также способствует феминизации этих женщин.
Психосексуальное и хирургическое лечение пациентов с синдромом частичной нечувствительности к андрогенам является чрезвычайно сложным и во многом зависит от представленного фенотипа. Остеопения считается поздним признаком синдрома нечувствительности к андрогенам.
Молекулярный анализ показал, что фенотип может частично зависеть от мозаичности гена рецептора андрогена в соматических кл. Данное открытие основано на случае пациента с генотипом 46,XY, с преждевременным стоп-кодоном в экзоне 1 гена андрогенного рецептора, при этом имеющего признаки вирилизации (лобковые волосы и увеличение клитора), что объяснялось открытием аллелей дикого типа при тщательном исследовании секвенирующего геля. Наличие мозаицизма смещает фенотип в сторону более значительной степени вирилизации, по сравнению с ожидаемой при только генотипе мутантного аллеля.
Генетическое консультирование в семьях с мутацией гена андрогенного рецептора затруднено. В дополнение к отсутствию корреляции генотипа и фенотипа в семьях высока частота (27%) вновь возникших мутаций.
Снижение уровня глобулина, связывающего половые гормоны, после введения экзогенного андрогена (станозолола) коррелирует с тяжестью дефекта рецептора и может стать полезным клиническим инструментом. Имеются сообщения об успешной терапии ЛП-андрогенами у пациентов с синдромом частичной нечувствительности к андрогенам и разл. мутациями андрогенного рецептора в ДНК-связывающем домене и лиганд-связывающем домене.
Также имеются сообщения о мутациях андрогенных рецепторов у пациентов со спинальной и бульбарной мышечной атрофией, у которых клинические проявления, в т.ч. атрофия яичек, бесплодие, гинекомастия и повышение уровней ЛГ, ФСГ и эстрадиола, обычно проявляются между 3-м и 5-м десятилетиями жизни. Мутации андрогенных рецепторов также были описаны у пациентов с раком предстательной железы.
г) Неустановленные причины. У др. мужчин с недостаточной вирилизацией и кариотипом XY отмечаются существенные различия наружных и внутренних половых органов и разл. степень развития пениса и производных мюллеровых протоков. Яички м.б. гистологически нормальными или рудиментарными, или м.б. только одно яичко. У до 50% детей с НФП 46,XY не выявляется ни одна известная причина заболевания. Некоторая степень неоднозначности половых органов сопутствует целому ряду хромосомных аберраций, которые всегда должны учитываться при проведении ДД, наиболее распространенным из которых является синдром 45,X/46,XY. Для установления факта мозаицизма может потребоваться кариотипирование нескольких тканей.
Др. сложные генетические синдромы, многие из которых являются результатом мутации в одном гене, сочетаются с разл. степенью неоднозначности гениталий, особенно у мужчин. Эти расстройства необходимо идентифицировать на основании сопутствующих экстрагенитальных пороков развития.
1. Синдром Смита-Лемли-Опица — это АуР-заболевание, вызванное мутациями в гене стерол-Δ7-редуктазы, расположенном на хромосоме 11q12-q13. Для него характерны пре- и постнатальная задержка роста, микроцефалия, птоз, вывернутые вперед ноздри, широкие альвеолярные отростки, синдактилия II и III пальцев стопы и тяжелые когнитивные нарушения. Его частота составляет 1:20 000-30 000 живорождений в популяциях североевропейского и центральноевропейского происхождения; 70% составляют мужчины.
Пациенты с мужским генотипом обычно имеют неопределенные гениталии, а иногда имеет место частичная смена пола с неоднозначностью женских гениталий или полная смена пола с женскими наружными гениталиями.
Производные мюллеровых протоков обычно отсутствуют. У пациентов с кариотипом 46,XX при данном заболевании половые органы имеют нормальное строение. Было выявлено два типа синдрома Смита-Лемли-Опица: классическая форма (тип I), описанная ранее, и акродисгенитальный синдром, обычно приводящий к смерти в течение одного года и сочетающийся с тяжелыми пороками развития, постаксиальной полидактилией и выраженным нарушением строения НПО (тип II). Стеноз привратника желудка сопутствует синдрому Смита-Лемли-Опица типа I, а болезнь Хиршспрунга — синдрому типа II. При синдроме типа II отмечались расщелина нёба, аномалии скелета, а в одном случае имелась липома гипофиза.
Некоторые авторы уверены в существовании разл. степеней тяжести этого заболевания, а не разл. заболеваний в выше представленной классификации.
Как при типе I, так и при типе II заболевания обнаруживается низкий уровень холестерина в плазме крови и повышенный уровень его предшественника — 7-дегидрохолестерина, при этом уровни этих в-в не коррелируют с тяжестью заболевания. С тяжестью заболевания, по-видимому, коррелирует уровень аполипопротеина Е у матери. Наиболее частым пренатальным проявлением синдрома Смита-Лемли-Опица является ЗВУР.
НФП 46,XY также было описано у братьев и сестер с синдромом α-талассемии и умственной отсталости.