Генотип — XX, половые железы представлены яичниками, однако наблюдается вирилизация НПО. Значительной пренатальной продукции АМГ не отмечается, т.к. половые железы представлены яичниками. Следовательно, развиваются матка, фаллопиевы трубы и шейка матки. Разновидностей и причин этого состояния относительно немного. Большинство случаев возникает вследствие в/утробного воздействия избыточного количества экзогенных или эндогенных андрогенов на плод женского пола.
Изменения заключаются главным образом в вирилизации НПО (гипертрофия клитора и сращение половых губ).
а) Врожденная дисфункция коры надпочечников. Это заболевание является наиболее частой причиной НПР с атипией половых органов и с кариотипом 46,XX. У детей женского пола с недостаточностью 21-гидроксилазы и 11-гидроксилазы наблюдается значительная вирилизация, хотя при недостаточности 3β-гидроксистероид-дегидрогеназы типа II также отмечается минимальная вирилизация (см. рис. ниже).
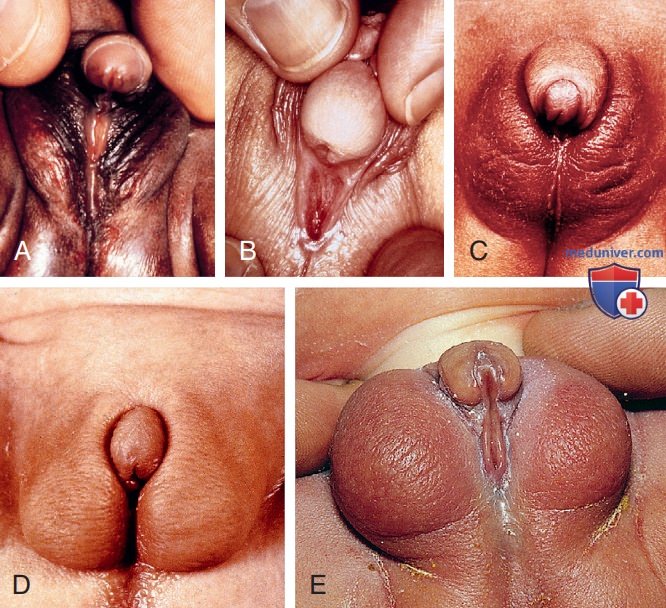
Примеры атипичных половых органов. Эти клинические случаи описывают овотестикулярное расстройство полового развития (A) и врожденную гиперплазию надпочечников с вирилизацией (B-E).
Пациентки с сольтеряющей формой врожденной гиперплазии надпочечников, обусловленной недостаточностью 21-гидроксилазы, как правило, сильнее вирилизованы, чем пациентки с врожденной гиперплазией коры надпочечников без потери соли. Маскулинизация м.б. настолько сильной, что образуется пенильная уретра, и пациентка может казаться мужчиной с двусторонним крипторхизмом.
б) Дефицит ароматазы. У женщин с кариотипом 46,XX редкое заболевание — дефицит ароматазы, в период в/утробного развития приводит к 46,XX НПР полового созревания из-за неспособности яичников синтезировать эстроген развивается гипергонадотропный гипогонадизм.
В качестве примера данного заболевания можно привести двух младенцев с кариотипом 46,XX, у которых при рождении отмечались увеличение клитора и сращение половых губ в задних отделах. В одном из этих случаев уровень эстрогена в сыворотке крови и моче матери был очень низким, а уровень андрогенов в сыворотке крови — высоким. Уровень эстрогена в сыворотке пуповинной крови также был крайне низким, а уровень андрогенов — повышенным.
У второй пациентки также отмечалась вирилизация неясной этиологии с рождения, однако недостаточность ароматазы не была диагностирована до 14 лет, когда у нее произошла дальнейшая вирилизация и не наступило половое созревание. На тот момент ее уровень гонадотропных гормонов и андрогенов был повышен, уровень эстрогенов — понижен, а УЗИ выявило кисты яичников больших размеров с обеих сторон. На примере этих двух пациенток показана важная роль ароматазы в превращении андрогенов в эстрогены.
Известны дополнительные случаи недостаточности ароматазы вследствие мутаций в гене ароматазы (CYP19) у пациентов женского и мужского пола. Были описаны клинические наблюдения двух родных сиблингов с дефектом данного гена: оба были высокого роста из-за отсутствия опосредованного эстрогенами закрытия эпифизарной зоны роста. Пробанд 28 лет, с кариотипом XX, была ростом 177,6 см (+2,5 SD) после лечения заместительной гормональной терапией. Ее 24-летний брат был ростом 204 см (+3,7 SD) и имел костный возраст = 14 годам.
Низкодозированная заместительная терапия эстрадиолом, тщательно подобранная с целью поддержания его нормального, соответствующего возрасту уровня, м.б. показана больным девушкам даже до наступления полового созревания.
в) Резистентность к кортизолу, обусловленная мутацией гена глюкокортикоидного рецептора. У 9-летней девочки с НПР 46,XX, которое, предположительно, было вызвано недостаточностью 21-гидроксилазы (врожденной гиперплазией надпочечников) с 5-летнего возраста, отмечались повышенный уровень кортизола, как в исходном состоянии, так и после дексаметазона, АГ и гипокалиемия, что указывает на предположительный диагноз генерализованной резистентности к ГКС.
Было продемонстрировано наличие неизвестной ранее гомозиготной мутации в экзоне 5 глюкокортикоидного рецептора. В данной бразильской семье заболевание было АуР. Вирилизация происходит из-за избыточной стимуляции синтеза надпочечниковых стероидов АКТГ, т.к. дефект глюкокортикоидных рецепторов присутствует и в гипофизе, который определяет неадекватный эффект кортизола для обеспечения «-» обратной связи.
1. Дефицит Р450-оксидоредуктазы. Цитохром Р450 оксидоредуктаза, кодируемая геном 7q11.2, является кофактором, необходимым для нормальной ферментативной активности микросомальных 21-и 17-гидроксилаз. Т.о., дефицит оксидоредуктазы вызывает частичные комбинированные стероидогенные дефекты Р450С17 и Р450С21. Девочки рождаются с неоднозначными половыми органами, но, в отличие от классической формы врожденной гиперплазии коры надпочечников, вирилизация не прогрессирует после рождения, а уровень андрогенов в норме или понижен. Мальчики могут родиться с недостаточной степенью вирилизации.
У детей обоих полов могут отмечаться костные аномалии, наблюдаемые при синдроме Энтли-Бикслера. И наоборот, в серии пациентов с синдромом Энтли-Бикслера у пациентов с неопределенными гениталиями и нарушенным синтезом стероидных гормонов наблюдался дефицит оксидоредуктазы. У пациентов без неопределенных половых органов с нормальным синтезом стероидных гормонов отмечались мутации рецептора 2 фактора роста фибробластов.
К главным отличительным чертам синдрома Энтли-Бикслера относятся краниосиностоз, тяжелая гипоплазия средней части лица, проптоз, атрезия/стеноз хоан, выступающие лобные бугры, диспластические уши, вдавленная переносица, лучеплечевой синостоз, переломы длинных костей и искривление бедренной кости, а также аномалии МПС.
г) Вирилизирующие опухоли у матери. В редких случаях плод женского пола подвергается вирилизации из-за материнской опухоли, секретирующей андрогены. В нескольких случаях образование было представлено доброкачественной аденомой надпочечников, однако во всех остальных случаях это были опухоли яичника, в частности андробластомы, лютеомы и опухоли Крукенберга (табл. 6). Вирилизация матери может проявляться увеличением клитора, угревой сыпью, снижением тона голоса, снижением лактации, гирсутизмом и повышением уровня андрогенов. У младенца наблюдается увеличение клитора разл. степени, часто со сращением половых губ.
Матерям детей с необъяснимыми НПР 46,XX необходимо провести физикальное обследование и исследование уровней тестостерона, дегидроэпиандростерона-сульфата и андростендиона в плазме.
д) Влияние лекарственных препаратов с андрогенным эффектом на беременных женщин. Сообщалось, что тестостерон и метилтестостерон в некоторых случаях вызывают развитие НПР 46,XX (см. табл. 6). Наибольшее число случаев было вызвано применением определенных гестагенных ЛП для лечения угрозы прерывания беременности. С тех пор эти гестагены были заменены на ЛП, не обладающие вирилизующим действием.
Сообщалось о младенцах с вирилизацией, кариотипом 46,XX и каудальными аномалиями, у которых вирилизирующий агент идентифицировать не удалось. В таких случаях расстройство обычно сочетается с др. врожденными дефектами, особенно МВП и ЖКТ. Последовательности ДНК, специфичные для У-хромосомы, в т.ч. SRY, отсутствуют. В одном случае был обнаружен шов мошонки и повышенный уровень тестостерона, однако причина осталась неизвестной.
1. Мутации SF-1. В ходе всемирного исследования пациентов с 46,XX овотестикулярным НПР была выявлена специфическая мутация в SF-1, p.Arg92Trp. Функциональные исследования показали, что продукт мутации, вероятно, вмешивался в ингибирование развития яичек. В одной семье с мутацией, передающейся по материнской линии, у матери наблюдалась ранняя менопауза. Сообщалось, что многие др. мутации SF-1 вызывают изолированную недостаточность яичников, некоторые из них ассоциированы с НПР 46,XY у их потомства.
2. Тестикулярное нарушение полового развития при кариотипе 46,XX. При этом состоянии, также известном как синдром XX у мужчин, половые железы представлены яичками, а вирилизация обычно неполная. После выхода из детского возраста могут развиться бесплодие и/или гонадная недостаточность. Многие случаи возникают в результате транслокации последовательности SRY на одну из Х-хромосом, часто в сочетании с дупликацией SOX-9. Могут возникнуть трудности с определением подходящего пола воспитания.
3. 46,XX дисгенезия гонад. Клиническая картина у этих девушек развивается в период полового созревания и характеризуется нормальными женскими гениталиями, отсутствием развития МЖ и гипергонадотропным гипогонадизмом. Структуры, в норме развивающиеся из мюллеровых протоков, определяются, но яичники отсутствуют или представлены в виде тяжа.
4. Неясного/неизвестного генеза. В редких случаях 46,XX НПР может сочетаться с др. врожденными аномалиями, особенно с пороками развития МВП или ЖКТ, и, следовательно, имеет многофакторное происхождение. К ним относятся клоакальная экстрофия и ассоциация MURCS (гипоплазия мюллеровых протоков, агенезия почек и аномалии развития шейно-грудных сомитов). Изолированный порок развития Мюллеровых протоков известен как синдром Майера-Рокитанского-Кюстера-Хаузера.