Синдром Харбойяна: врожденная дистрофия роговицы и глухота
Комбинация врожденной дистрофии роговицы с прогрессирующей нейросенсорной глухотой описана у 2 из 10 епбеов, рожденных от одного двоюродного брака, и у 1 из 10 сибсов, рожденных от другого двоюродного брака того же самого отца (Harboyan et al.).
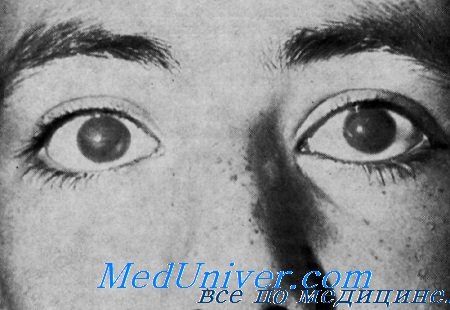
Орган зрения. С рождения отмечалась белесоватая мутность роговицы со снижением остроты зрения. Офтальмологическое исследование 3 больных в возрасте от 12 до 50 лет обнаружило такие же изменения. Эпителий роговицы был шероховатым, по не был окрашен. Строма ее была отечной, утолщенной и гомогенно белой. Острота зрения у 12-летнего мальчика на оба глаза была снижена до 20/200. У мужчин 28 и 50 лет счет пальцев был возможен только на расстоянии мене 1 м. У женщин 28 и 50 лет счет пальцев также был доступен только на расстоянии менее 1 м.
У молодых женщин было обнаружено повышение внутриглазного давления, тогда как у пожилых женщин, после наступления менопаузы, отмечалось ухудшение зрения. Эти наблюдения позволяют предположить некоторое прогрессирование нарушений зрения.
Орган слуха. Потеря слуха была впервые отмечена в возрасте от 10 до 25 лет. В дальнейшем она медленно прогресировала. На аудиограмме у 12-летнего мальчика была обнаружена двусторонняя нейросеисорная глухота в диапазоне от 20 до 50 дБ, выраженная более резко на высоких частотах. У 2 его полусибсов (сестер) потеря слуха достигала 40—70 дБ. Различение речи составляло 90—100%, SISI и tonedecay-тесты были отрицательными.
Вестибулярная система. У всех 3 больных калорические вестибулярные пробы были нормальными.
Лабораторные данные. Рутинные лабораторные исследования, включая определение уровня мукополисахаридов в моче, были нормальными.
Наследственность. Здоровые родители и существование кровного родства между ними ясно указывают на то, что синдром наследуется по аутосомно-рецессивному типу.
Диагноз. Врожденная дистрофия роговицы может быть изолированной патологией, наследующейся по аутосомно-рсцессивному типу (Маumenee). Помутнение роговицы может быть симптомом некоторых мукополисахаридозов (синдром Гурлер, Шейе и Марото— Лами). Эти заболевания, однако, клинически вполне различимы. При врожденной глаукоме встречаются помутнение роговицы, светобоязнь, расширение роговицы, увеличение внутриглазного давления и слезотечение. При синдроме Когана отмечаются светобоязнь и инъецирование глаз, а кератит расположен глубоко в строме.
При синдроме Когана отмечаются также шум в ушах, резкое головокружение и, наконец, выраженная глухота с утратой вестибулярной функции. Необходимо также исключить врожденный сифилитический кератит.
При аутосомно-рецессивной дистрофии роговицы Фера (Fehr) изменения роговицы становятся очевидными в течение первого десятилетия жизни, при настоящем же синдроме имеется врожденная дистрофия роговицы (Francois). Мы осведомлены только об одном случае сочетания дистрофии роговицы Фера с врожденной невральной глухотой. Больной был рожден от кровно-родственного брака (Моrо, Ameidi).
Другим возможным примером синдрома Харбойяна являются сибсы, описанные Scialfa с сотр. Родители были кровными родственниками. Однако у этих сибсов наблюдались умственная отсталость и клинодактилия V пальцев.
Лечение. Показаны трансплантация роговицы и лечение глаукомы. Могут быть использованы слуховые аппараты.
Прогноз. Потеря зрения и слуха медленно прогрессирует.
Выводы. Главные черты синдрома: 1) аутосомно-рецессивное наследование; 2) врожденная дистрофия роговицы с тенденцией к медленному прогрессированию; 3) выявляющаяся в детстве, медленно прогрессирующая нейросенсорная глухота.