Глухота с чашеобразными ушами и слезно-ушным-зубо-пальцевым синдромом
Hollister, Klein, De lager, Lachman и Rirhoin сообщили о мексиканской семье, в которой у отца и 5 из 8 его детей выявили повое заболевание, обозначив его как слезно-ушной-зубо-пальцевой синдром (лакримо-аурикуло-денто-дигитальный синдром).
Клинические данные. Данные осмотра. У всех больных, кроме одного, выявлены непроходимость слезного канала и гипоплазия слезной точки. Этот дефект проявлялся хроническим слезотечением, случаями гноетечения, дакриоциститом и конъюнктивитами в раннем детстве.
Аномалии пальцев были различными: конусовидные II и III пальцы, конусовидные и/или удвоенные концевые фаланги I пальца руки, клинодактилия V пальцев, гипоплазия I пальца и синдактилия II и III пальцев.
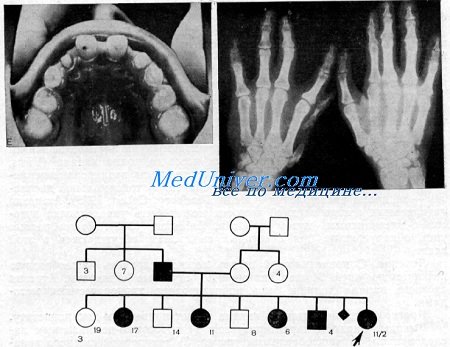
Зубы были мелкими с гипопластичной эмалью. Коронки латеральных резцов на верхней челюсти имели коническую форму.
Орган слуха. Уши у всех больных были чашеобразной формы. У некоторых из них отмечалась резко выраженная проводящая или нейросенсорная глухота, у других была выявлена легкая высоко- или низкочастотная нейросенсорная потеря слуха пли дефект звукопроводимости. Ригидность проводящего аппарата, установленная посредством импеданс-аудиометрии, позволила предположить отосклероз или аномалии слуховых косточек.
Вестибулярная система. Об исследовании вестибулярной функции не упоминается.
Коронки латеральных резцов верхней челюсти конической формы. Малые коренные зубы ненормально заострены и выглядят чрезмерно стертыми.
Рентгенография рук, показанных на рисунке копчике правого большого пальца выражен дополнительный центр окостенения. На левой руке отмечается гипоплазия большой многоугольной кости и фаланг большого пальца. Пястье представлено двумя широко разделенными центрами окостенения.
Родословная пораженной семьи.
Лабораторные данные. Рентгенограммы. Рентгенография рук выявила широкую вариабельность костных аномалий. Томограммы были плохо выполнены.
Другие исследования. У тех же самых сибсов была выявлена односторонняя гипоплазия или аплазия почки.
Патология. Исследование височных костей не проводилось.
Наследственность. Синдром наследуется по аутосомно-доминаитпому типу.
Диагноз. Закупорка слезного канала встречается у 1—6% нормальных новорожденных. Он становится проходимым на 3-м месяце жизни в результате спонтанного прорывания носового конца капала (Viers). Отсутствие слезной точки может являться изолированным феноменом или одним из симптомов эктродактилоэктодермальной дисплазии — синдрома расщепления (Gorlin, Pindborg, Cohen). Агенезия латеральных резцов верхней челюсти может быть унаследована аутосомно-доминантным путем (Gorlin, Goldman).
Чашеобразные уши, обнаруженные при некоторых синдромах, обсуждаются в этой статье.
Лечение. С косметической целью можно исправить чашеобразную деформацию ушей. Можно применять слуховые аппараты. Атрезия слезного канала должна быть корригирована хирургическими методами.
Прогноз. Атрезия слезного капала может привести к дакриоциститу.
Выводы. Синдром характеризуется: 1) аутосомно-доминантным наследованием;
2) чашеобразными ушами;
3) закупоркой слезного канала и гипоплазией слезной точки; 4) различными аномалиями пальцев;
5) конической формой коронок латеральных резцов верхней челюсти;
6) несовершенным образованием зубной эмали;
7) смешанной глухотой.