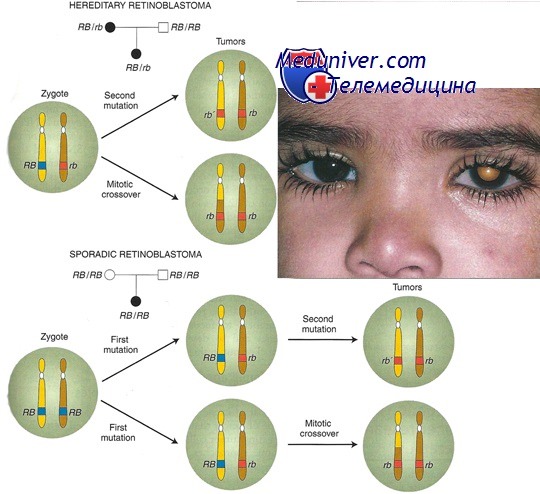
Ретинобластома, образец болезни, вызываемой мутацией в гене-супрессоре опухолевого роста, — редкая злокачественная опухоль сетчатки у младенцев, со встречаемостью около 1 на 20 000 новорожденных.
Диагноз ретинобластомы обычно предполагает удаление пораженного глаза, хотя небольшие, рано диагностированные опухоли можно лечить местной терапией с целью сохранения зрения.
Около 40% случаев ретинобластомы — наследственная форма, при которой ребенок наследует один мутантный аллель в локусе ретинобластомы (RB1). К развитию опухоли приводят соматические мутации или другие изменения в одной единственной клетке сетчатки, приводящие к утрате функции оставшегося нормального аллеля.
Заболевание наследуется как доминантный признак, поскольку появление соматической мутации весьма вероятно из-за большого числа первичных ретинобластов (более 106) и их быстрой пролиферации. Поскольку шанс «второго удара» при наследственной форме столь велик, патологические изменения часто происходят более чем в одной клетке, и, таким образом, гетерозиготы по заболеванию часто поражаются множественными опухолями, нередко обоих глаз.
С другой стороны, «второй удар» не происходит в 100% случаев, являясь случайным событием; следовательно, пенетрантность ретинобластомы, хотя и высокая, но не полная.
Оставшиеся 60% случаев ретинобластомы ненаследственные (спорадические); в этих случаях оба аллеля RB1 в одной клетке сетчатки инактивировались независимо друг от друга. Поскольку два таких события в одной клетке — редкий случай, обычно наблюдают только моноклональную опухоль и только в одном глазу.
Хотя спорадическая ретинобластома обычно происходит только в одном участке одного глаза, у 15% больных с односторонней ретинобластомой имеется наследственный тип со случайным развитием опухоли только в одном глазу. Другое различие между наследственными и спорадическими опухолями состоит в том, что средний возраст начала спорадической формы — ранний дошкольный возраст, позже, чем у детей с наследуемой формой.
Генетика ретинобластомы
Потеря гетерозиготности при ретинобластоме
Генетики, изучавшие полиморфизмы ДНК в области, близкой к локусу RB1, сделали необычное, но очень значимое генетическое открытие, когда проанализировали аллели, встречающиеся в опухолевых тканях у пациентов с ретинобластомой. Больные с ретинобластомой, гетерозиготные по полиморфным локусам вблизи гена RB1 в нормальных тканях, например в лейкоцитах, имели опухоль, содержащую аллели только из одного гомолога хромосомы 13 с потерей гетерозиготности (loss of heterozygositi — LOH) в регионе гена.
В семейных случаях маркеры на хромосоме 13 оказались унаследованными от больного родителя, т.е. с одним аномальным аллелем RB1. Таким образом, потеря гетерозиготности представляет собой «второй удар» по оставшемуся аллелю. Потеря гетерозиготности может происходить вследствие интерстициальной делеции, но есть и другие механизмы, например митотическая рекомбинация или нерасхождение хромосом. Потеря гетерозиготности — наиболее общий мутационный механизм нарушения функции нормального аллеля RB1 у гетерозигот.
Когда потеря гетерозигоности не выявляется, «второй удар» обычно — соматическая мутация второго гена или случайная транскрипционная инактивация нормального аллеля метилированием. Потеря гетерозиготности характерна для множества других опухолей, как наследуемых, так и спорадических, и часто считается подтверждением существования гена-супрессора опухолевого роста, даже если этот ген неизвестен.
Ген RB1 картирован на хромосоме 13, в положении 13ql4. У небольшого процента больных с ретинобластомой первая мутация — цитогенетически обнаруживаемая делеция или транслокация этой части хромосомы 13. Подобные хромосомные изменения, если они также нарушают смежные с RB1 гены, могут вызывать, кроме ретинобластомы, дополнительные дисморфические симптомы.
Выжившие дети с наследственной формой ретинобластомы имеют существенно повышенный (в 400 раз) риск развития других опухолей в последующем. Риск значительно выше, если ребенок получал лучевую терапию, поскольку пациенты с наследственной формой уже несут мутацию в одном аллеле RB1 во всех клетках их тела и, следовательно, подвержены другим опухолям, если теряют вторую копию.
Мезенхимальные опухоли, обычно развивающиеся в зрелости, включают остеогенные саркомы, фибросаркомы и меланомы. Хотя ген RB1 экспрессируется во многих тканях, потеря RB1 вызывает опухоли в детстве только в сетчатке и, позже в жизни, в тканях мезенхимального происхождения. Причина тканевой специфичности неизвестна.
Продукт гена RB1, названный p110 Rbl (белок размером 110 килодальтон), — фосфопротеин, сначала гипофосфорилируемый, а затем гиперфосфорилируемый на разных этапах клеточного цикла. В гипофосфорилированном состоянии он блокирует клеточный цикл между G1 и S, тормозя вход в S-фазу, за счет связывания и инактивации фактора транскрипции, обеспечивающих синтез ДНК.
Когда белок p110 Rbl становится более фосфорилированным, он высвобождает факторы транскрипции, допуская вход клетки в S-фазу; затем последовательно дефосфорилируется в течение клеточного цикла, что позволяет ему снова функционировать, блокируя переход в S-фазу следующего цикла. Утрата гена RB1 лишает клетки важной митотической контрольной точки и приводит к неконтролируемой пролиферации. Следовательно, ген RB1 — образец гена-супрессора опухолевого роста типа ХКЦ. Известно, что ген RB1 видоизменяется во многих клеточных линиях, происходящих из определенных различных опухолей в ходе их формирования.