Атаксия Фридрейха, или спиноцеребеллярная атаксия, представляет четвертую категорию болезней тринуклеотидных повторов. Болезнь наследуется по аутосомно-рецессивному типу, в отличие от болезни Гентингтона, миотонической дистрофии и синдрома ломкой Х-хромосомы.
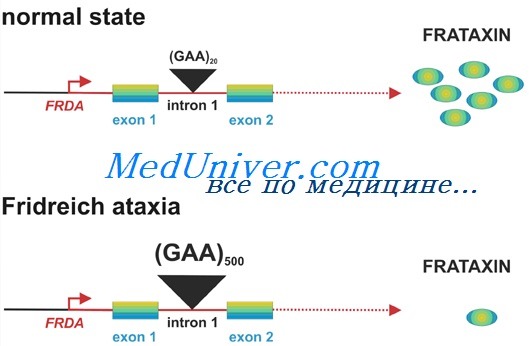
Заболевание, обычно обнаруживаемое в юности, характеризуется несогласованностью движений конечностей, затруднениями речи, сниженными или отсутствующими сухожильными рефлексами, снижением стереотактического и вибрационного чувств, кардиомиопатией, сколиозом и аномалией стоп. В большинстве случаев атаксия Фридрейха вызвана экспансией совершенно другого тринуклеотидного повтора — AAG, располагающегося на этот раз в интроне гена, кодирующего митохондриальный белок фратаксин, участвующий в метаболизме железа.
У здоровых число повторов изменяется от 7 до 34 копий, у больных обычно от 100 до 1200 копий. Экспансия в пределах интрона создает помехи нормальной экспрессии гена фратаксина. Поскольку атаксия Фридрейха рецессивная, чтобы вызвать болезнь, необходима недостаточная экспрессия обоих аллелей. Фактически 1-2% пациентов с атаксией Фридрейха — компаундные гетерозиготы, с одним аллелем — частой мутацией интрона за счет повторов AAG и вторым — другой нуклеотидной мутацией.
Сравнение болезни Гентингтона (и других нейродегенеративных полиглутаминовых болезней) с синдромом ломкой Х-хромосомы, миотонической дистрофией и атаксией Фридрейха показывает некоторое сходство, но также и множество различий. Хотя экспансия нестабильных тринуклеотидных повторов представлена во всех четырех типах болезней, экспансия при полиглутаминовых болезнях происходит в кодирующем регионе и содержит от 40 до 120 копий CAG, а повторы при синдроме ломкой Х-хромосомы, миотонической дистрофии и атаксии Фридрейха включают триплеты с другими нуклеотидами, содержат сотни тысяч повторяющихся триплетов и располагаются в нетранслируемых участках генов FMR1, DPPK и FRDA соответственно.
Премутации, вызывающие повышение риска экспансии в полную мутацию характерны для всех четырех заболеваний, а в родословных доминантных и Х-сцепленных болезней (болезнь Гентингтона, синдром ломкой Х-хромосомы и миотоническая дистрофия) отмечают антиципацию. В то же время количество повторов в аллелях премутации при болезни Гентингтона от 29 до 35, как и при миотонической дистрофии, что значительно меньше, чем при синдроме ломкой Х-хромосомы.
У носителей премутации может развиться клинически значимое заболевание при синдроме ломкой Х-хромосомы, но они здоровы при болезни Гентингтона и миотонической дистрофии. Экспансия аллелей премутации происходит преимущественно у женщин при атаксии Фридрейха, миотонической дистрофии и синдроме ломкой Х-хромосомы; самые крупные экспансии, вызывающие раннее начало болезни Гентингтона, происходят в мужских половых клетках.
Наконец, степень митотической нестабильности при синдроме ломкой Х-хромосомы, миотонической дистрофии и атаксии Фридрейха много больше, чем при болезни Гентингтона, и приводит к значительно большей изменчивости в количестве повторов, обнаруживаемых среди клеток той же самой ткани и между другими соматическими тканями у одного больного.