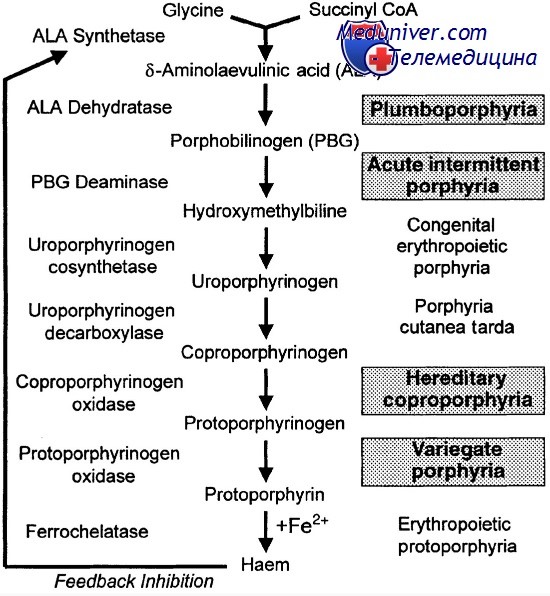
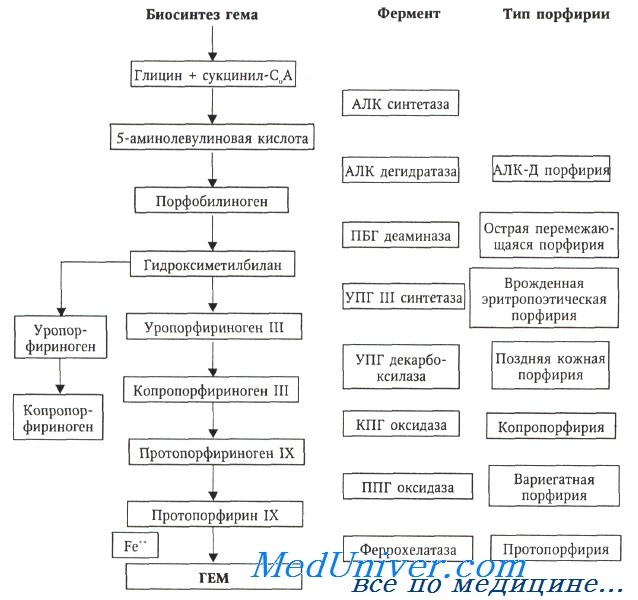
Порфирия - это метаболическое заболевание, при котором нарушается тот или иной этап синтезирования конечного продукта — протопорфирина. Независимо от наследственного или приобретенного характера болезни, порфирия отличается неполноценностью функции отдельных ферментов на определенной стадии процесса синтезирования протопорфирина. При наследственной порфирпи причину следует искать в мутации, происходящей в структурном или регулирующем гене, обусловливающем синтез данного фермента, в то время как при приобретенной порфирии функцию, качественно нормально синтезирующего фермента нарушает какое-либо токсическое вещество (свинец, пестицид и пр.).
Даже при наследственной порфирии отдельные, инородные для организма вещества (например, барбитураты, фенилбутазон и пр.), могут взаимодействовать с метаболическими функциями ферментативной цепи, которая, до той поры, была более или менее компенсированной (Гайдош, Петрилэ и сотр.). В зависимости от локализации недостатка в процессе образования протопорфирина различаются:
а) предпироловые расстройства, когда молекула не синтезирует пироловое ядро (дельта-аминолевулиновую кислоту);
б) монопироловые расстройства, наблюдаемые при возникновении препятствий в образовании порфибилкногена;
в) тетрапироловые расстройства, при которых не осуществляется декарбоксилирование боковых радикалов пирола (уро-, копро- и протопорфирин), равно как и определение типа изомерии.
В любом случае аномалия передается по наследству аутосомальным путем. В некоторых случаях наличие отягчения требуется у обоих родителей (рецессивная передача), как, например, при врожденной эритропоэтической порфирии, в то время как в других — лишь у одного из них (доминантная передача), например при острей перемежающейся порфирии. Опубликованные до настоящего времени сведения об эритропоэтических порфириях (копро- и протопорфирии) еще скудны, способ передачи не уточнен, однако важная роль видимо отведена фактору «проникновения» отягчения.
Поздняя кожная порфирия бывает наследственной, симптоматической или вторичной.
Для большинства этих нозологических единиц уточнен метаболический недостаток. Так, при врожденной эритропоэтической порфирии нарушается ряд этапов метаболического процесса, из них наиболее важным является сокращение активности изомеразы, которая оказывается неспособной снабжать, посредством изомера III, первым порфирином, синтезируемым после слияния четырех молекул порфобилиногена (с помощью порфобилиногендезаминазы). Это способствует чрезмерному скоплению патологического уропорфирина.
Тем не менее синтезирование нормального порфирина не нарушается, в связи с этим гем вырабатывается в нормальном количестве и развитие анемии это результат дальнейшего вторичного расплавления крови за счет переизбытка аномальных метаболитов (отсутствие фактора, угнетающего репрессорные гены).
Для личных нужд клеток организма необходимы разные количества продуктов с геминным ядром. Пироловые продукты организма в подавляющем большинстве сопряжены с белками и лишь небольшие их количества несвязанны. Хотя эти продукты не рассматриваются как собственно токсические для клеток, однако их скопление за определенные пределы оказывает влияние на отдельные метаболические процессы клеток.
При наследственной порфирии мутации, происходящие в этих процессах синтезирования включены в генетический код каждой клетки организма, независимо от выполняемой ею функции (у большинство клеток эти гены находятся в репрессированном состоянии). Наибольшую долю молекул стетрапироловым ядром синтезируют эритробласты и гепатоциты.
Часть избыточных продуктов обмена поступает в общее кровообращение, откуда удаляются с выделениями, в то время как другая (не удаленная) — скопляется во всех клетках организма. Однако не все клетки одинаково переносят эти продукты. Так, некоторые из них, в том числе невроны, кожные клетки и пр. наиболее чувствительны и в связи с этим — подобно скоплению железа при гемохроматозе — скопление продуктов порфирина обусловливает определенные селективные клинические проявления.
Порфириновые метаболиты вызывают две основные категории вредных последствий — демиэлинизацию и фотодерматоз. Поражение неврона (демиэлинизация) обусловливает не только нейропсихические явления, но также брюшные (рези, запор, рвоту и пр.). Скопление в коже некоторых из отмеченных соединений вызывает разряд энергии при контакте с ультрафиолетовым светом; в результате отмечаются светохимические реакции, ведущие к разрушению лизосом и дальнейшему выделению кислых гидролаз, определяющих кожные поражения.
На порфириновом синдроме можно доказать взаимное потенцирование наследственных и средовых факторов. Взаимодействие биологического фонда порфирина и средовых факторов или метаболических нарушений различной природы (цирроз печени, расстройство нервного характера и пр.) создает условия, при которых, компенсированные до той поры клинические проявления порфирина, становятся явными за счет истощения компенсирующих рессурсов.