Нервная система при нарушении синтеза и транспорта креатинина
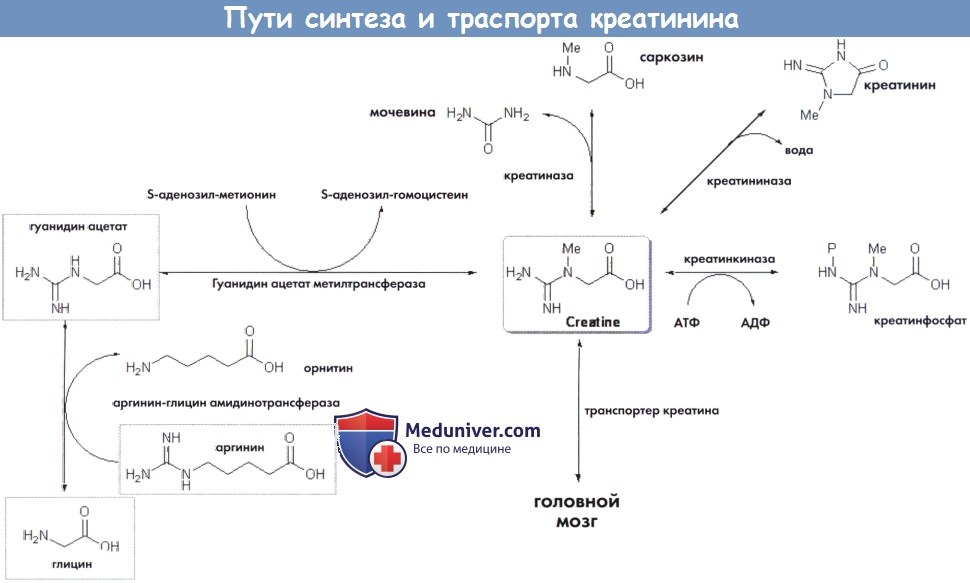
Креатинин и креатинфосфат являются основными субстратами накопления и переноса энергии. Креатинин синтезируется преимущественно в печени и поджелудочной железы в рамках двух-ступенчатого механизма из глицина, аргинина и метионина и в итоге путем неферментной циклизации превращается в креатинин.
В организме человека запас креатина поддерживается за счет биосинтеза и поступления с пищей по 1-2 г в сутки. Креатин переносится кровью, а на уровне клеток — креатинин-транспортирующей системой.
В течение последних нескольких лет у человека были идентифицированы врожденные нарушения метаболизма всех трех белковых компонентов.
а) Дефицит гуанидиноацетат-метилтрансферазы (GAMT). Первый пациент с таким заболеванием был зарегистрирован Stockier et al. (1994). Ребенок родился здоровым, но в дальнейшем у него развился гипотонус, экстрапирамидные дистонические симптомы и тяжелая задержка психомоторного развития. На МРТ, выполненной в возрасте 12 месяцев, выявлен аномальный сигнал от бледного шара.
При протонной МР спектроскопии выявлено отсутствие сигналов креатина и креатинфосфата в головном мозге, что предполагает врожденное нарушение синтеза креатина. Дефицит активности GAMT выявлен в ткани печени. В ряде случаев дефицит GAMT может быть выявлен при протонной МР спектроскопии как отсутствие сигнала креатинина/фосфокреатинина в головном мозге (von Figura et al., 2000). Поддерживающее применение креатинин-моногирата (4 г в сутки) приводит к заметному улучшению и нормализации аномалий скорлупы, выявляемых при МР-спекртроскопии.
Тем не менее, концентрация креатина/креатинфосфата в головном мозге, оцениваемая при МР-спектроскопии, не достигает нормальных значений. В возрасте 15 лет пациент был способен самостоятельно ходить, но у него отмечалась тяжелая задержка умственного развития с аутистическим самодеструктивным поведением.
Дефицит GAMT наследуется аутосомным путем, ген расположен на хромосоме 19р.13.3. Выявлено несколько мутантных аллелей дефицита GAMT. Клинические проявления заболевания различны и включают мышечную гипотонию, дистонию, тяжелую эпилепсию и задержку умственного развития.
Диагностику дефицита креатина следует проводить у пациентов с гипотоническими-дистоническими симптомами в сочетании с аномалиями на МРТ или без них. Патогномоничные лабораторные изменения при дефиците GAMT включают снижение концентрации креатина и уровня креатинина в крови, спинномозговой жидкости и моче, в то время как уровень гуанидинацетата (предшественника креатина) в крови, моче и спинномозговой жидкости повышен.
Отсутствие сигнала креатина/креатинфосфата по результатам МР-спектроскопии головного мозга достаточно для постановки диагноза. Дальнейшие биохимические и генетические исследования позволяют идентифицировать лежащее в основе нарушение.
Пути синтеза и транспорта креатинина.
б) Дефицит аргинин: глицин амидинотрансферазы (AGAT). При дефиците AGAT, впервые описанном в 2000 г., отклонений на МРТ головного мозга не выявляется, но при МР-спектроскопии обнаруживается полное отсутствие креатина/креатинфосфата. Диагностика дефицита AGAT основана на исследовании фибробластов и лимфоцитов.
У пациентов не отмечалось эпилепсии или других нейромышечных симптомов. Уровень креатина в сыворотке не отличался от нормы, уровень GAA был несколько снижен, а экскреция GAA с мочой — чрезвычайно снижена. У обоих пациентов отмечалась хорошая реакция на лечение креатином (Schulze, 2003).
в) Сцепленный с Х-хромосомой дефицит транспортера креатина (CRT1). Данное заболевание проявляется легкой задержкой умственного развития и тяжелой задержкой развития речи у мальчиков. Не считая легкой гипотонии, двигательные функции не нарушены. При МРТ и МР-спектроскопии отмечается практически полное отсутствие сигнала креатина.
Данное наследственное нарушение метаболизма вызвано генетическим дефектом сцепленного с Х-хромосомой транспортера креатина, локализованного на участке SLC6A8 хромосомы Xq28 (Salomons et al., 2003). Обнаружены не связанные между собой семьи, в которых выявлены мужчины и женщины носители дефектного гена. Клиническими проявлениями дефицита CrT1 является умеренная мышечная гипотония, задержка умственного развития, задержка развития речи и эпилепсия. Концентрация креатина в плазме и моче повышена, уровень GAA не отличается от нормы.
МР-спектроскопия является наиболее целесообразным неинвазивным исследованием. Пероральное применение креатина не проводило к восстановлению нормальной концентрации креатина в головном мозге (по результатам МР-спектроскопии) и не уменьшало выраженность симптомов (Schulze, 2003).