Нервная система при болезни Вольмана и болезни накопления эфиров холестерина (БНЭХ)
Болезнь Вольмана и болезнь накопления эфиров холестерина (БНЭХ) являются двумя фенотипическими вариантами экспрессирования дефицита лизосомальной кислой липазы (ЛАЛ), который приводит к выраженному накоплению эфиров холестерина и триглицеридов в большинстве тканей. Болезнь Вольмана представляет собой рано начинающуюся форму заболевания, всегда смертельную, в то время как БНЭХ является более доброкачественным вариантом с поздним началом (Assmann и Seedorf, 1995).
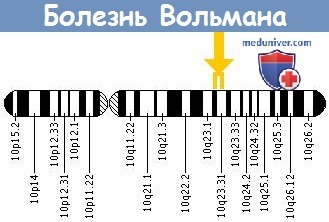
Оба заболевания наследуются аутосомно-рецессивным путем. В настоящее время идентифицированы структурные мутации гена LAL, локализованного на 10 хромосоме (10q23.2—23.3), без явной корреляции генотипа и фенотипа (Muntoni et al., 1995).
Возможна пренатальная диагностика путем оценки активности ЛАЛ в амниотических клетках или образцах ворсин хориона.
Клинические проявления включают тяжелую диарею, гепатомегалию и недостаточную прибавку в весе. Гипотония и задержка умственного развития проявляются после исходно нормального развития. Для болезни Вольмана характерна выраженная кальцификация надпочечников.
Диагностика основана на выявлении дефицита ЛАЛ в культуре фибробластов, лимфоцитах или других тканях. Гиперлипопротеинемия типична для БНЭХ и отсутствует при болезни Вольмана.
При болезни Вольмана специфическое лечение отсутствует. Комбинированное лечение холестирамином и ингибитором HMG-кофермент А редуктазы, корректирующее дислипопротеинемию при БНЭХ, никогда не применялось при болезни Вольмана. Трансплантация печени успешно проводилась у пациентов с БНЭХ с тяжелым фиброзом печени (Leone et al., 1995).