Болезни Пелицеуса-Мерцбахера и Александера у детей
Болезнь Пелицеуса-Мерцбахера представляет собой группу заболеваний, для которых характерны нистагм и аномальное строение миелина. Классическая форма заболевания, передающаяся по Х-сцепленному рецессивному типу наследования, вызвана аномалиями гена, кодирующего синтез протеолипидного белка, необходимого для формирования миелина в ЦНС и дифференциации олигодендроцитов. Мутации в том же гене могут привести к развитию семейного спастического парапареза.
Болезнь Пелицеуса-Мерцбахера характеризуется нистагмом и блуждающими движениями глаз в сочетании с кивательными движениями головы в младенческом возрасте. Ген локализован на хромосоме Xq22. Разработана молекулярная диагностика болезни Пелицеуса-Мерцбахера с использованием мутационного анализа. Однако, как и при большинстве заболеваний с Х-сцепленным типом наследования, молекулярная диагностика болезни Пелицеуса-Мерцбахера вызывает ряд затруднений, так как мутации в экзонах выявляются только у 10-25 % пациентов с этим заболеванием.
У детей с болезнью Пелицеуса-Мерцбахера отмечаются задержка развития, атаксия, хореоатетоз и спастичность, а также атрофия зрительных нервов и дизартрия. Заболевание заканчивается летальным исходом на 2-м или 3-м десятилетии жизни. Основные патоморфологические изменения включают отсутствие миелиновой оболочки неповрежденных аксонов, что предполагает нарушение функции олигодендроглии. Исследования показали существование генетического дефекта в биосинтезе протеолипида апопротеина — белка, играющего роль в дифференциации и поддержании функции олигодендроцитов.
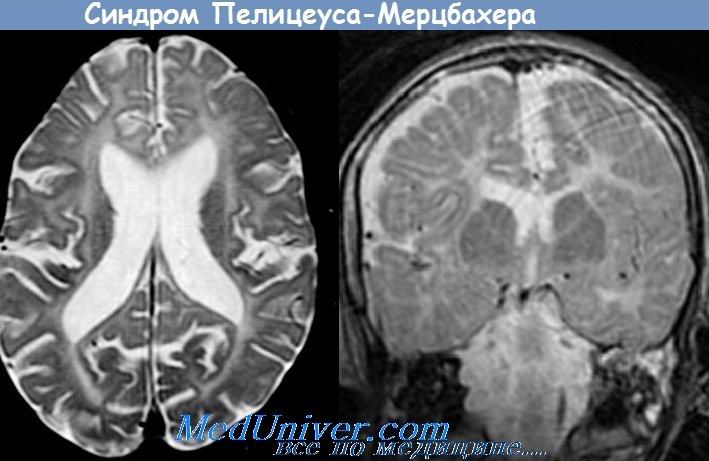
На МРТ — симметричный паттерн замедления миелинизации. Исследования слуховых ВП ствола на ранних этапах заболевания демонстрируют паттерн отсутствия волн III—V. Эти данные имеют большое значение при обследовании мальчиков раннего возраста с нистагмом. При исследовании ЗВП отмечается замедление латентностей, ССВП — отсутствие кортикальных ответов или замедление латентностей.
Болезнь Александера у детей
Болезнь Александера редкое спорадическое заболевание с прогрессирующей макроцефалией на первом году жизни. Заболевание обусловлено мутациями в гене глиального фибриллярного кислого протеина (GFAP). Патоморфологическое исследование ткани мозга выявляет отложения эозинофильных гиалиновых телец в периваскулярных пространствах мозга и в пределах мягкой оболочки мозга.
Дегенерация белого вещества наиболее выражена в лобных долях; на КТ в этой стадии отмечается соответствующее снижение плотности белого вещества мозга. У детей с болезнью Александера развиваются прогрессирующее снижение интеллекта, спастичность и эпилептические приступы, резистентные к антиэпилептической терапии; летальный исход наступает к 5 годам жизни.