Агенезия и дисгенезия мозолистого тела - клиника, диагностика
Агенезия и дисгенезия мозолистого тела Может быть полной или частичной. В последних случаях задняя часть обычно утрачена, из-за того, что мозолистое тело развивается в переднезаднем направлении, тем не менее, возможна и передняя агенезия (Aicardi et al., 1987; Barkovich и Norman, 1988b; Sztriha, 2005). Встречаются атипические формы, сложно отличимые от голопрозэнцефалии (Barkovich, 1990). Агенезия мозолистого тела относительно распространена. Распространенность среди населения в целом оценивается как 3-7/1000 (Bedeschi et al., 2006).
Наблюдаемая частота возросла с введением в практику КТ и MPT. Jeret et al. (1987) выявили 33 случая в серии 1447 КТ снимков.
Даже теперь диагноз в основном ставится только пациентам с неврологическими симптомами, поэтому истинная частота не известна.
Отсутствие мозолистой спайки, как правило, замещается двумя продольными связками, известными как продольное мозолистое тело или пучки Пробста, проходящие по внутренней стороне полушарий. Часто появляются борозды на внутренней стороне с радиальным расположением и расширением затылочных рогов, сохраняющих их фетальную морфологию, так называемую кольпоцефалию (Noorani et al., 1988). Другие связанные аномалии часто включают в себя формирование кист кзади от третьего желудочка, сообщающиеся или несообщающиеся (Yokota et al., 1984, Griebel et al., 1995, Barkovich et al., 2001b) аномалии червя мозжечка и ствола мозга, а также смешанные мальформации ЦНС, такие как гетеротопия, аномалии образования извилин или цефалоцеле (Jeret et al., 1987; Barkovich и Norman, 1988b; Serur et al., 1988).
Гигантские кисты могут иметь благоприятный исход, несмотря на внушительные размеры (Lena et al., 1995; Haverkamp et al, 2002). Пороки развития ЦНС были обнаружены у 33% пациентов с полной и у 42% из них с частичной агенезией (Bedeschi et al., 2006).
Именно эти сочетанные аномалии отвечают за клинические проявления. Липома мозолистого тела почти неизменно сопровождает агенезию этой структуры (Zee et al., 1981; Vade и Horowitz, 1992). Характерны периферические мальформации (Parrish et al., 1979). Особенно часто встречаются глазные аномалии (Aicardi et al., 1987). Гипоплазия мозолистого тела (Bodensteiner et al., 1994) может быть минимальной формой каллозной дисгенезии, но гораздо чаще следствием кортикальных нейрональных потерь.
Этиология разнообразна. Идентифицировано не менее 46 синдромных пороков развития или метаболических расстройств и 30 мутантных генов (Kamnasaran, 2005). При несиндромных формах, генетическая передача встречается редко, хотя имеются сообщения о семейных случаях с аутосомно-рецессивным (Finlay et al., 2000), Х-сцепленным рецессивным (Menkes et al., 1964; Kaplan, 1983) и аутосомно-доминантным типом наследования (Aicardi et al. 1987). Выявлено множество хромосомных дефектов, включая трисомию 8, 13, 16 и 18, а также смесь менее распространенных хромосомных дефектов.
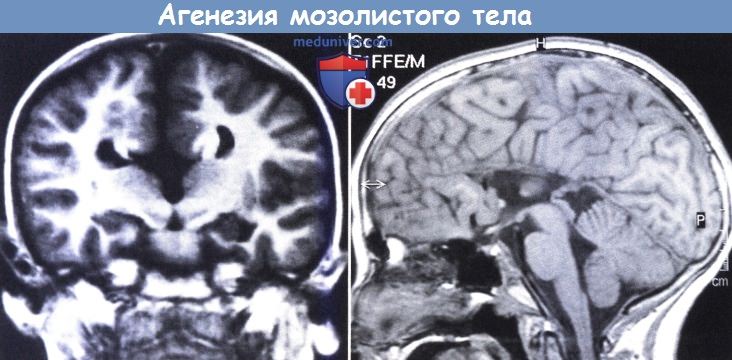
Агенезия мозолистого тела, (слева) МРТ (инверсия-восстановление):
продольное мозолистое тело (пучки Пробста) около внутренней поверхности тел желудочков мозга.
(справа) Сагиттальная проекция: полное отсутствие мозолистого тела и радиальное распределение борозд на внутренней стороне полушарий.
Serur et al. (1988) просмотрели 81 случай из литературных источников, из которых в 21 была трисомия 8, у 14 была трисомия 13-15 и у 18 затронуты хромосомы 17 или 18. Из 34 выполненных кариотипов в двух была обнаружена трисомия 8. Субтеломерные абберации были выявлены в 5% случаев Bedeschi et al. (2006). Среди экологических факторов выделяют плодный алкогольный синдром. Некоторые метаболические заболевания, особенно гипергликемия (Dobyns, 1989), недостаточность пируватдегидрогеназы (Bamforth et al., 1988, Raoul et al., 2003) и другие метаболические расстройства вместе составляют около 2% случаев агенезии мозолистого тела. В большинстве случаев их происхождение неизвестно.
Описание клинических проявлений каллозной аге-незии можно разделить на две части: несиндромные и синдромные формы (Davila-Guttierez, 2002).
Несиндромные формы наиболее распространены (Jeret et al., 1987; Serur et al., 1988). Неизвестный процент случаев остается бессимптомным или случайно выявляется только благодаря большим размерам головы. У большинства пациентов отмечается задержка умственного развития, судороги и/или большие размеры головы (Aicardi et al., 1987). Часто обнаруживаются гипертелоризм. В исследовании Jeret et al. (1987) 82% пациентов имели умственную отсталость или задержку развития, 43% страдали судорогами и у 31% развился церебральный паралич.
Однако нормальное когнитивное развитие наблюдалось у 9 из 63 детей (Bedeschi et al., 2006), возможно, и чаще, поскольку бессимптомные случаи, вероятно, не диагностированы. Возможны судороги любого типа, включая инфантильные спазмы, но чаще — очаговые. Хотя характерно увеличение размеров головы, иногда более 5-7 СО от среднего, показания к шунтированию достаточно строги, так как многие случаи «гидроцефалии» спонтанно стабилизируются, не причиняя каких-либо проблем. Макроцефалия может быть частично связана с наличием гигантских кист, расположенных кзади от третьего желудочка (Barkovich et al., 2001b).
Специфические расстройства межполушарной передачи либо отсутствуют, либо только минимальные (Jeeves и Temple, 1987).
Тем не менее, имеются сообщения о тонких нарушениях межполушарной связи и топографической памяти. В редких случаях может наблюдаться эндокринологическая патология (Paul et al., 2003).
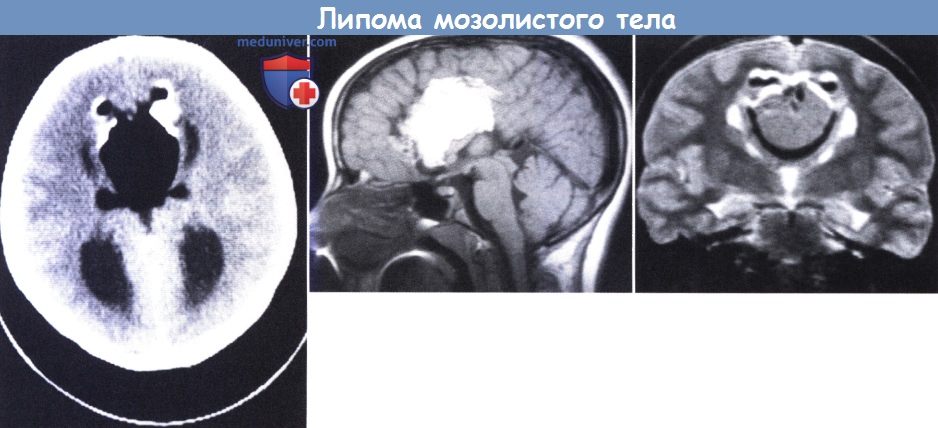
Липома мозолистого тела у 8-летней девочки с парциальным комплексом судорог, но без неврологических нарушений.
(слева) КТ: крупные массы жировой плотности, разделенные передними рогами боковых желудочков, с периферической кальцификацией и двумя небольшими латеральными участками распространения жировых масс.
(в центре) МРТ (сагиттальная проекция): замещение мозолистого тела тканью липомы.
(справа) МРТ (Т 2-взвешенная последовательность): полное замещение каллезного тела жировой тканью.
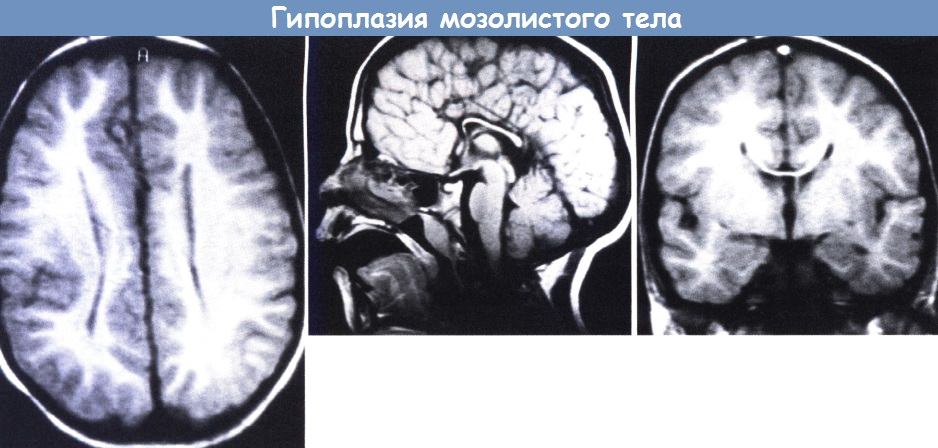
Гипоплазия мозолистого тела, (слева) МРТ (аксиальная проекция): вид желудочков в соответствии с агенезией мозолистого тела.
(в центре) Сагиттальная проекция: полное мозолистое тело с коленом и валиком, но укороченное и истонченное. Обратите внимание на радиальное расположение медиальной извилины.
(справа) Фронтальная проекция: широкое разделение тел желудочков пролабирующей лимбической извилиной.
Синдромные формы перечислены в таблице ниже.
Синдром Айкарди (Chevrie и Aicardi, 1986, Aicardi, 2005) имеет отношение примерно к 1% случаев с инфантильными спазмами, вероятно, из-за Х-сцепленных доминантных мутаций. Встречается почти исключительно у девочек, хотя имеется сообщение о двух случаях у мальчиков с XXY набором хромосом. Известно только об одном семейном случае у двух сестер (Molina et al., 1989). Характерные особенности синдрома включают в себя инфантильные спазмы и специфические хориоидальные лакуны, часто в сочетании с колобомой зрительного диска. Позвоночно-реберные аномалии имеются в половине случаев. Исход обычно неблагоприятный, с сохраняющимися судорогами и глубокой умственной отсталостью. Спектр тяжести оказался шире, чем предполагалось ранее (Menezes et al, 1994). В редких случаях может присутствовать мозолистое тело (Aicardi, 1994, 1996).
Диагноз определяют хориоидальные лакуны и сопутствующие аномалии, выявленные при МРТ (перивентрикулярная гетеротопия, диспластичная кора, эпендимальные кисты). При патологическом исследовании в мозге обнаруживают многочисленные участки гетеротопии и полимикрогирии не разделенного на слои типа (Billette de Villemeur et al., 1992), тогда как так называемые лакуны представляют собой истончение пигментного эпителиального и сосудистого слоя с утратой пигментных гранул. Эпендимальные кисты часто обнаруживают вокруг третьего желудочка. Кисты или опухоли сосудистого сплетения могут достигать больших размеров (Aicardi, 2005). При выявлении вместе с агенезией мозолистого тела возможен пренатальный диагноз.
Другие синдромные формы встречаются редко или в большинстве ограничены определенными этническими группами.
Синдром Айкарди у трехмесячной девочки.
Обратите внимание на асимметрию полушарий, двусторонние кисты сосудистого сплетения, кисты вокруг третьего желудочка с различным сигналом от цереброспинальной жидкости и перивентрикулярные гетеротопии.
Эпендимальное происхождение кист сосудистого сплетения было подтверждено при гистологическом исследовании.
Агенезия мозолистого тепа. Ультразвуковое антенатальное исследование, сагиттальная проекция.
Обратите внимание на нормальный четвертый желудочек, отсутствие эхосигнала от мозолистого тела и расширенный боковой желудочек.
Слева на фотографии — затылок плода.
Семейный синдром агенезии мозолистого тела с патологией гениталий, который также может проявляться с микроцефалией и другими аномалиями ЦНС, является частью более обширного спектра расстройств, связанных с мутациями в гене ARX на хромосоме Хр22.3 (Hartmann et al, 2004).
Синдром Андерманна был описан у французского канадца в районе озера Сент-Джонс (Andermann, 1981), но о нескольких случаях было заявлено за пределами Канады. Этот синдром затрагивает периферическую нервную систему в дополнение к агенезии мозолистого тела или гипотрофии. Агенезия мозолистого тела зачастую является частью рото-лице-пальцевого синдрома I типа.
Синдром периодической гипотермии и потливости (Shapiro et al., 1969) можно эффективно лечить с помощью клонидина, который был испытан в связи с открытием изменений метаболизма норадреналина при этом синдроме. Правда, половина случаев не сопровождается каллозной агенезией (Sheth et al., 1994). Имеются сообщения об агенезии мозолистого тела с периодической гипертермией («обратный синдром Шапиро») (Hirayama et al., 1994).
Диагноз агенезии каллозного тела основывается на данных нейровизуализации. Диагноз полной агенезии несложен при ультрасонографии, КТ и MPT (Aicardi et al., 1987; Jeret et al., 1987; Serur et al., 1988). MPT эффективнее при диагностике частичной агенезии. Диагностика методами нейровизуализации не представляет сложностей, при КТ или МРТ выявляется подъем третьего желудочка и широкое разделение передних рогов с классической картиной «бычьих рогов» на фронтальных срезах. Диффузионно-тензорная МР (Lee et al., 2005) позволяет выявить отклонение трактов, особенно пучков Пробста, которые направляются кзади около стенки желудочка и не пересекают противоположную сторону.
Пренатальная диагностика возможна с 22 недели (Bennett et al., 1996; Simon et al., 2000a). Решение о прерывании беременности сложно принять безоговорочно, пока нет данных о распространенности бессимптомных случаев. Blum et al. (1990) сообщали, что у 6 из 12 новорожденных, у которых агенезия мозолистого тела была диагностирована антенатально, имели нормальное развитие в возрасте 2-8 лет. Moutard et al. (2003) наблюдали 17 случаев с пренатально диагностированной изолированной агенезией с повторными измерениями IQ. В возрасте 6 лет все дети имели коэффициент умственного развития на нормальном уровне с тенденцией к нижней границе нормы. Девять детей в исследовании Bedeschi et al. (2006) имели нормальное развитие.
Тем не менее, у 2 из 9 детей, пренатально диагностированных с помощью МРТ без сочетанных аномалий, отмечалась задержка в развитии (Volpe et al., 2006). Случаи, связанные с другими мальформациями или хромосомными аномалиями, неизменно имели неблагоприятный исход. Поэтому крайне важно фетальное кариотипирование и полное обследование на наличие сочетанных пороков развития.