Наследственные спастические параплегии. Болезни с вовлечением нервной системы
Дегенеративные заболевания ЦНС с преимущественным вовлечением пирамидной системы являются генетически гетерогенными и могут наследоваться по аутосомно-доминантному, аутосомно-рецессивному и X-сцепленному рецессивному типам [Behan W., Maia M., 1974; Harding A., 1984; Reid E., 1999]. Их распространенность в европейских популяциях составляет около 10 на 100 000 [Polo J. et al., 1991].
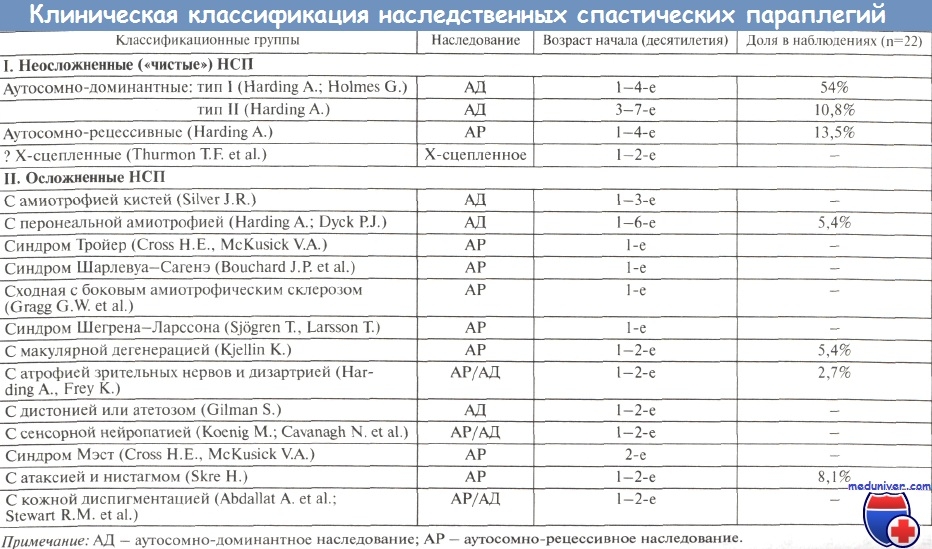
Изолированная форма спастической параплегии является наиболее частой и в большинстве случаев наследуется по аутосомно-доминантному типу. Рецессивным типам наследования чаще соответствуют различные осложненные варианты заболевания.
Для аутосомно-доминантных генетических вариантов наследственной спастической параплегии характерна широкая вариабельность возраста дебюта клинической симптоматики - от первого до седьмого десятилетия жизни. Появление указанных дополнительных симптомов (которые обычно являются негрубыми и выявляются после многолетнего течения болезни) не противоречит диагнозу «чистой» спастической параплегии. В отдельных семьях описаны осложненные формы аутосомно-доминантной спастической параплегии, когда у больных нижний спастический парапарез сочетается с дегенерацией сетчатки, атрофией зрительных нервов, сенсорной невропатией, амиотрофиями, экстрапирамидными расстройствами, деменцией, эпилептическими припадками, нарушением пигментации кожи и др. [Harding A., 1984; Reid E., 1999].
Течение заболевания медленно прогрессирующее, серьезная инвалидизация наступает через 15-20 лет от момента появления первых субъективных жалоб. На секции выявляется дегенерация пирамидных путей в боковых столбах спинного мозга (особенно в каудальных отделах), изменения обнаруживаются также в задних столбах, спиноцеребеллярных и переднем пирамидном трактах.
В соответствии с современной номенклатурой, различные локусы и соответствующие им генетические варианты наследственных спастических параплегии обозначаются символами SPG1, SPG2 и т.д. (от англ. Spastic Paraplegia Gene). К настоящему времени известны 8 самостоятельных генетических вариантов аутосомно-доминантных спастических параплегии, сцепленных со следующими хромосомами: 14ql 1.2-24.3 (SPG3); 2р21-24 (SPG4); 15qll.l (SPG6); 8q24 (SPG8); 10q23.3-24.2 (SPG9); 12ql3 (SPG10); 19ql3.1 (SPG12) и 2q24-34 (SPG13) [Ilazan J. et al., 1993; 1994; Fink J. et al., 1995; lledera P. et al., 1999; Reid E. et al., 1999; 2000; Sen M. et al., 1999; Fontaine B. et al., 2000].
В ряде семей локализация мутантного гена с известными хромосомными локусами исключена [Reid E. et al., 1999], что свидетельствует о существовании еще одного или нескольких, пока не установленных генетических вариантов доминантных параплегии.
Анализ клинико-генетических корреляций в семьях с аутосомно-доминантными спастическими параплегиями в целом показал отсутствие каких-либо существенных фенотипических различий между отдельными формами: в абсолютном большинстве семей, независимо от хромосомного локуса, имеет место клиническая картина «чистой» спастической параплегии. Специфический осложненный фенотип характерен лишь для формы SPG9: у этих иольных нижний спастический парапарез сочетается с катарактой, гастроэзофагеальным рефлюксом и амиотрофиями [Seri M. et al., 1999]. Определенной особенностью формы SPG3 является более ранний возраст манифестации симптомов болезни (в среднем в обследованных семьях заболевание проявлялось в возрасте < 10 лет).