Генетические болезни почек - кратко с точки зрения внутренних болезней
Появление современных генетических методов, таких как секвенирование нового поколения, позволило значительно расширить наши знания в этой области и получить гораздо более полное представление о наследственных заболеваниях почек.
а) Наследственные гломерулопатии:
1. Синдром Альпорта. Наследственные гломерулопатии представлены несколькими орфанными заболеваниями детского возраста, а среди взрослой популяции самым частым генетически детерминированным заболеванием этой группы является синдром Альпорта.
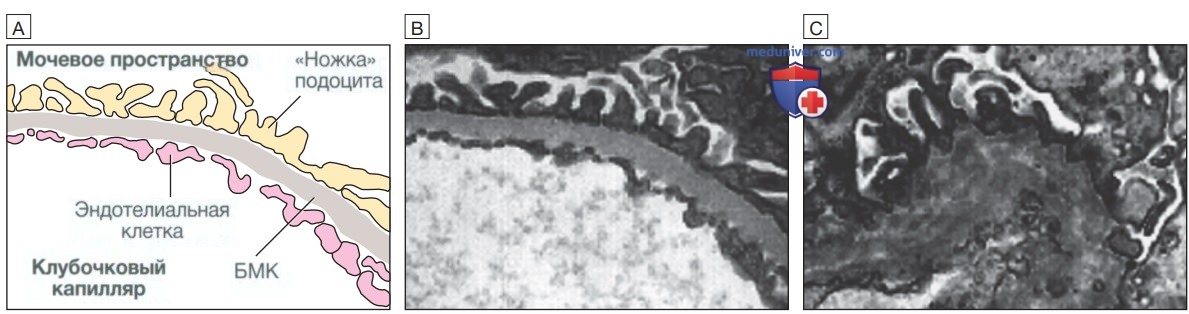
Большинство случаев болезни обусловлены мутацией или делецией в гене COL4A5, который кодирует синтез коллагена IV типа и располагается на Х-хромосоме. Тип наследования этого самого распространенного варианта синдрома Альпорта — Х-сцепленный рецессивный. Менее распространенные варианты заболевания с аутосомно-рецессивным типом наследования обусловлены мутациями в генах COL4A3 или COL4A4, расположенных на хромосоме 2. Аккумуляция патологического коллагена в БМК приводит к ее прогрессирующей дегенерации (рис. 1), клинические проявления которой варьируют от изолированной гематурии до ТПН, развивающейся в старшем подростковом или юношеском возрасте.
Рисунок 1. Синдром Альпорта: А — схематическое изображение базальной мембраны клубочков (БМК) в норме; В — нормальная базальная мембрана клубочков (электронная микрофотография) содержит в основном тканеспецифичные для почек (αЗ, α4 и α5) цепи коллагена IV типа; С — при синдроме Альпорта структура базальной мембраны клубочков нарушается за счет замещения цепей α3, α4 и α5 цепями α1 и α2. Хотя в раннем возрасте базальная мембрана клубочков представляется структурно неизмененной, со временем развиваются ее истончение, расщепление и дегенерация, которые в конечном итоге приводят к значительному утолщению базальной мембраны клубочков.
У женщин носительство мутаций в гене COL4A5 обычно ограничивается фенотипом с редким развитием тяжелого повреждения почек, основным клиническим проявлением которого является гематурия. Аналогичным образом могут поражаться и другие базальные мембраны, в состав которых входят те же изоформы коллагена, что особенно относится к улитке, поэтому синдром Альпорта сопровождается развитием нейросенсорной тугоухости и патологией со стороны органов зрения.
Ингибиторы АПФ могут замедлять, но не предотвращать прогрессирующее снижение почечных функций. Пациенты с синдромом Альпорта являются подходящими кандидатами для ЗПТ, поскольку они молоды и, как правило, не имеют сопутствующих заболеваний. Однако у очень небольшого числа пациентов, подвергшихся трансплантации почки, может развиваться иммунная реакция на антигены коллагена БМК донорской почки, что приводит к гибели аллотрансплантата.
2. Болезнь тонких базальных мембран. Клиническими проявлениями болезни тонких базальных мембран, как правило, являются микрогематурия клубочкового происхождения, не сопровождающаяся АГ, протеинурией и снижением СКФ. При световой микроскопии не удается выявить каких-либо изменений и клубочки кажутся абсолютно нормальными, однако электронная микроскопия позволяет обнаружить патологическое истончение БМК.
Болезнь может носить семейный характер, и у некоторых пациентов выявляется носительство мутаций в генах синдрома Альпорта, однако, по-видимому, это справедливо не для всех случаев, и у многих пациентов причина заболевания остается неясной. В связи с тем что у некоторых пациентов с течением времени отмечается появление протеинурии, которая в долгосрочной перспективе может быть ассоциирована с увеличением риска прогрессирования ХБП, целесообразно осуществлять длительное наблюдение таких пациентов.
3. Наследственный нефротический синдром. К настоящему времени уже известно много генов, ответственных за развитие нефротического синдрома в молодом возрасте, гистологическая картина которого часто соответствует ФСГС. Наследование этих генов не сцеплено с полом и может быть аутосомно-доминантным или аутосомно-рецессивным. Следует отметить, что аутосомно-доминантному типу соответствует более благоприятный фенотип с неполной пенетрантностью и отсроченным дебютом.
Почти все гены, ассоциированные с развитием нефротического синдрома, кодируют белки подоцитов, включая нефрин (нефропатия финского типа) и подоцин, — в обоих случаях развивается ранний врожденный нефротический синдром. Кроме этого, ФСГС, будучи следствием различных аутосомно-доминантных мутаций, может быть частью некоторых синдромов, таких как болезнь Шарко—Мари—Тута (ген INF2), синдром ногтей-надколенника, или наследственная артроостеоониходисплазия (ген LMX1B), и сочетание патологии половых органов, опухоли Вильмса и умственной отсталости (ген WTI).
Наследование вариантного гена APOL1, которое наблюдается преимущественно у людей западноафриканского происхождения, приводит к значительному увеличению риска развития ФСГС.
б) Наследственные тубулоинтерстициальные заболевания. В последние годы стало очевидным, что значительное количество случаев ХБП, при которых протеинурия либо совсем отсутствует, либо умеренно выражена, являются генетически детерминированными заболеваниями и наследуются по аутосомнодоминантному или аутосомно-рецессивному типу (табл. 20). Эта группа достаточно гетерогенна, но их гистологические паттерны идентичны другим формам хронического тубулоинтерстициального нефрита.
Несмотря на то что преобладающим морфологическим субстратом этих заболеваний является картина тубулоинтерстициального нефрита, в связи с наличием небольших кист, нередко обнаруживаемых в паренхиме мозгового вещества почки, применительно к ним широко использовался термин «медуллярная кистозная болезнь почек». Многие из этих заболеваний, особенно те, которые ранее назывались нефронофтизом, сопровождаются дистрофией сетчатки и аномалиями головного мозга или других органов, а некоторые могут быть связаны с гиперурикемией или подагрой (мутации UMOD или HNF1-β). Достижения современной генетики позволили внести некоторую терминологическую и классификационную ясность в отношении этой многочисленной группы заболеваний.
в) Изолированные нарушения функции канальцев. К настоящему времени описано достаточно большое количество патологических состояний, в основе которых лежат специфические дефекты молекул-транспортеров, экспрессированных в клетках канальцевого эпителия, однако здесь упомянуты только самые распространенные из них. Почечная глюкозурия представляет собой доброкачественное нарушение канальцевой реабсорбции глюкозы вследствие мутаций в гене натрийглюкозного котранспортера 2-го типа (SGLT2) с аутосомнорецессивным типом наследования, при котором глюкозурия наблюдается у лиц с неповышенной концентрацией глюкозы в плазме крови.
На основе этих данных были созданы и активно используются в лечении сахарного диабета ингибиторы SGLT2, и на сегодняшний день уже имеются данные об их способности улучшать как почечные, так и сердечно-сосудистые исходы.
Цистинурия — редкая патология, вследствие которой в канальцах нарушается реабсорбция профильтровавшихся в клубочках молекул — цистина, орнитина, аргинина и лизина. Причиной заболевания являются мутации в гене переносчика аминокислот SLC3A1. Высокая концентрация цистина в моче может приводить к образованию цистиновых камней.
К другим редким генетически детерминированным тубулопатиям относят наследственный гипофосфатемический рахит, при котором снижается реабсорбция профильтрованного клубочками фосфата; нефрогенный несахарный диабет, возникающий вследствие резистентности канальцев к действию вазопрессина (антидиуретического гормона); а также синдромы Барттера и Жительмана, при которых развиваются гипонатриемия и гипокалиемия вследствие повышенной экскреции этих микроэлементов с мочой.
Синдром Фанкони представляет собой генерализованную проксимальную канальцевую дисфункцию. Это состояние обычно проявляется низкими концентрациями фосфатов и мочевой кислоты в крови, глюкозурией, аминоацидурией и проксимальным канальцевым ацидозом. В дополнение к причинам интерстициального нефрита, описанным выше, некоторые врожденные метаболические нарушения также могут быть ассоциированы с синдромом Фанкони, в частности, к ним относят болезнь Вильсона, цистиноз и наследственную непереносимость фруктозы.
Почечный канальцевый ацидоз является терминальной стадией различных тубулопатий с утратой функций дистальных (классический или тип 1) или проксимальных (тип 2) почечных канальцев.
г) Кистозные болезни почек. Достаточно часто в качестве случайной находки у пациентов обнаруживается одна или даже несколько кист в почках, особенно если возраст пациентов превышает 50 лет. Как правило, эти кисты бессимптомны и не оказывают никакого влияния на дальнейший прогноз, однако иногда они могут стать причиной появления болевого синдрома или гематурии. Вместе с этим для некоторых заболеваний установлена причинно-следственная связь с формированием множественных кист почек. Более подробно эти случаи обсуждаются ниже.
1. Поликистоз почек у взрослых. ПКП у взрослых — распространенное генетически детерминированное заболевание с аутосомнодоминантным типом наследования, распространенность которого составляет приблизительно 1:1000. Мелкие кисты, выстланные канальцевым эпителием, начинают развиваться с младенческого или детского возраста, и их увеличение происходит медленно и неравномерно. Увеличиваясь в размерах, кисты сдавливают и повреждают окружающую здоровую почечную паренхиму. До 85% случаев ПКП развивается вследствие мутации в гене PKD1, причиной оставшихся примерно 15% случаев заболевания является мутация в гене PKD2 (гены PKD1 и PKD2 кодируют белки полицистин 1 и полицистин 2 соответственно).
Средний возраст развития ТПН у пациентов с мутациями PKD1, которая формируется примерно у 50% пациентов, составляет 52 года. Среди меньшинства, у которых ПКП обусловлен мутациями в гене PKD2, средний возраст формирования ТПН составил 69 лет. По существующим на сегодняшний день оценкам, от 5 до 10% пациентов, получающих ЗПТ, страдают ПКП.
- Клиническая картина. Характерные клинические признаки перечислены в табл. 21. Обычно заболевание протекает бессимптомно вплоть до пожилого возраста, однако АГ нередко манифестирует уже с 20-летнего возраста. При осмотре пациента можно пропальпировать узловатую поверхность одной или обеих почек. Примерно в 30% случаев у пациентов с ПКП выявляются кисты печени, но нарушение ее функции при этом встречается редко. В некоторых случаях (почти всегда у женщин) поликистоз печени приводит к развитию тяжелой симптомной гепатомегалии, обычно сопутствующей увеличению почек, однако в ряде случаев поражение почек может быть незначительным.
Кроме этого, поликистозная болезнь почек в 5% случаев ассоциирована с развитием мешотчатых аневризм сосудов головного мозга, также называемых ягодными. Создается впечатление, что такая ассоциация в значительной степени ограничена отдельными семьями и, предположительно, обусловлена специфическими мутациями. Достаточно часто у больных ПКП может выявляться митральная или аортальная регургитация, которые редко бывают гемодинамически значимыми и достигают степени тяжелой клапанной недостаточности. Также нередко у этой категории пациентов обнаруживаются дивертикулы толстой кишки и грыжи брюшной стенки.
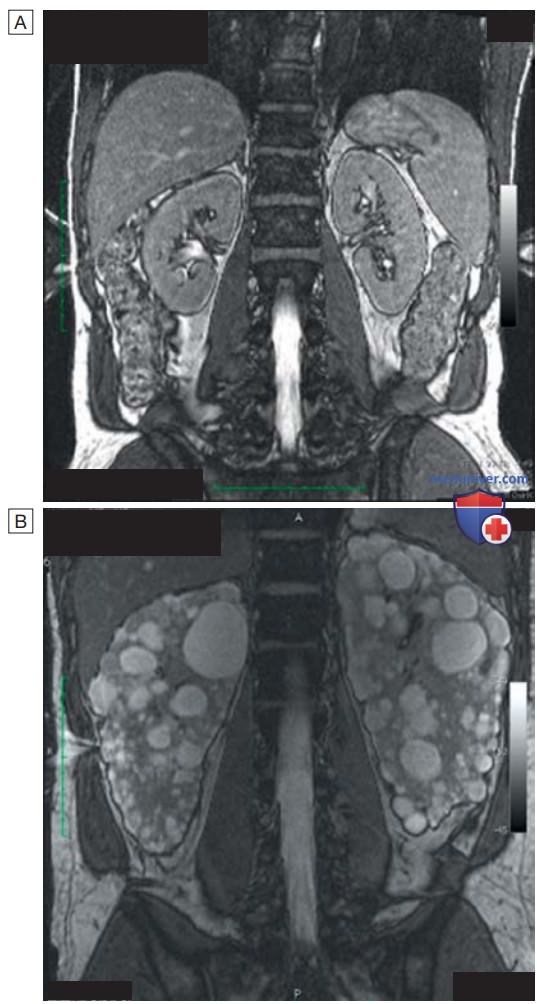
- Лабораторные и инструментальные исследования. Диагноз ставится на основании анализа данных семейного анамнеза, клинических проявлений и результатов УЗИ. УЗИ почек позволяет выявить кисты примерно у 95% пациентов в возрасте старше 20 лет и является предпочтительным методом исследования при подозрении на наличие ПКП, однако ограниченная разрешающая способность не всегда позволяет обнаружить мелкие развивающиеся кисты у более молодых пациентов. Помимо УЗИ, для диагностики кистозного поражения почек могут быть использованы и другие визуализирующие методы, например МРТ (рис. 2). Простые почечные кисты бывают и у здоровых людей, но они редко выявляются в возрасте до 30 лет.
Рисунок 2. Магнитно-резонансная томография почек: А — здоровые почки; B — поликистоз почек. Несмотря на значительное увеличение почек, у пациента лишь незначительно снижена скорость клубочковой фильтрации
На сегодняшний день для ультразвуковой диагностики ПКП у пациентов с неизвестным генотипом и положительным семейным анамнезом используются следующие диагностические критерии:
• 15—39 лет — не менее трех одно- или двусторонних кист почек;
• 40-59 лет — не менее двух кист в каждой почке;
• 60 лет и старше — не менее четырех кист в каждой почке.
Диагноз ПКП можно верифицировать посредством скрининга мутаций PKD1 или PKD2, однако метод традиционной секвенации в клинической практике используется редко, что обусловлено не только большими размерами гена PKD1, но и значительным количеством возможных мутаций. На сегодняшний день быстрее и проще генетический скрининг на PKD1 и PKD2 позволяет выполнить секвенирование нового поколения, проведение которого может быть информативно в сомнительных случаях (молодые пациенты, мало кист, отсутствие семейного анамнеза), а также для обследования живых доноров почек и для выявления мутаций, связанных с неблагоприятным прогнозом (см. ниже).
Скрининг на выявление внутричерепных аневризм при ПКП не предусмотрен, но возможно выполнение магнитно-резонансной ангиографии в семьях, имеющих отягощенный анамнез по субарахноидальным кровоизлияниям. Выявляемость интракраниальных аневризм вследствие такого скринирования низкая, но в то же время и соотношение пользы и риска от интервенционного вмешательства при бессимптомных аневризмах сосудов головного мозга у больных ПКП не определено.
- Лечение. У пациентов с болезнями почек в связи с наличием у них повышенного риска сердечно-сосудистой заболеваемости и смертности очень важно контролировать уровень АД на целевом уровне. Однако на сегодняшний день отсутствуют сведения о том, что достижение и поддержание у пациентов с ПКП целевых для ХБП значений АД (например, менее 130/80 мм рт.ст.) позволяет улучшить у них почечные исходы. В то же время было продемонстрировано, что достижение очень низких значений АД (менее 110/75 мм рт.ст.) на фоне терапии ингибиторами АПФ или БРА II приводило к замедлению скорости увеличения почек в объеме, однако не замедляло скорость снижения рСКФ, кроме того, переносимость такого уровня АД была плохой.
С другой стороны, именно поддержание таких жестких целевых значений АД, действительно, приводило к более выраженному снижению индекса массы миокарда ЛЖ, что может иметь значение для улучшения сердечно-сосудистого риска в более позднем возрасте.
Антагонист вазопрессиновых V2-рецепторов толваптан способен тормозить увеличение почек в объеме и скорость снижения СКФ. В настоящее время он зарегистрирован уже во многих странах для лечения пациентов с высоким риском прогрессирования ПКП (в России не зарегистрирован). К факторам риска прогрессирования относят крупные почки (объем почек, скорректированный по росту), усеченные мутации гена PKD1 и семейный анамнез прогрессирования в молодом возрасте, а также мужской пол, АГ, протеинурию и развитие ранних симптоматических кист.
Как правило, пациенты с ПКП являются подходящими кандидатами для проведения диализа и трансплантации. Иногда почки бывают настолько большие, что приходится удалять одну или сразу обе, чтобы освободить место для трансплантата. В остальных случаях почки удаляют только тогда, когда они являются источником инфекции или причиной болевого синдрома.
2. Другие кистозные заболевания почек. Мутации в гене HNF1-β приводят к развитию синдрома, включающего развитие сахарного диабета и кистозного поражения почек (см. выше), однако фенотип поражения почек весьма вариабелен и может быть представлен не только кистозной трансформацией, но и поражением тубулоинтер-стиция или врожденным отсутствием почки. На сегодняшний день этот синдром рассматривается в рамках одного из вариантов MODY.
Аутосомно-рецессивный ПКП, обусловленный мутациями в гене PKHD1, кодирующем синтез белка фиброцистина, встречается значительно реже, чем аутосомно-доминантный вариант поликистоза взрослых (около 1 на 20 000 живорожденных). Заболевание часто манифестирует в младенчестве или в раннем детском возрасте и проявляется кистозным поражением почек и врожденным фиброзом печени.
Некоторые орфанные наследственные заболевания с аутосомно-доминантным типом наследования ассоциированы с формированием в почках множественных кист или опухолей во взрослом возрасте. Так, например, при туберозном склерозе происходит замещение почечной паренхимы множественными ангиомиолипомами, что может иногда становиться причиной развития почечной недостаточности у взрослых. Помимо нефроангиомиолипоматоза, у этой категории больных повышен риск развития почечных кист и почечноклеточной карциномы.
Внепочечные проявления туберозного склероза включают поражения кожи (аденомы сальных желез на лице) и головного мозга (судороги и умственная отсталость). Синдром фон Гиппеля—Линдау характеризуется поражением почек, которое может быть представлено множественными почечными кистами, аденомами и даже аденокарциномой, а также экстраренальными проявлениями — вовлечением центральной нервной системы (гемангиобластомы), поджелудочной железы (серозные цистаденомы) и надпочечников (феохромоцитома).
К настоящему времени идентифицированы и другие, менее распространенные наследственные кистозные заболевания почек, которые имеют некоторое сходство с ПКП, но иные генетические причины развития. Поликистозная дисплазия почек чаще бывает односторонней и выявляется в детском возрасте, причем в большинстве случаев, по мере роста ребенка, только одна пораженная почка подвергается инволютивным изменениям, что становится причиной наличия единственной функционирующей почки во взрослом возрасте.
У пациентов, длительно страдающих почечной недостаточностью, может развиваться приобретенный ПКП, который не является наследственным, ассоциирован с повышенным синтезом эритропоэтина, а иногда и с развитием почечноклеточной карциномы.