Сурфактант представляет собой смесь фосфолипидов и белков, которые синтезируют и секретируют альвеолоциты II типа, выстилающие дистальные участки воздушного пространства. Эта смесь образует монослой на границе раздела воздух-жидкость, снижая поверхностное натяжение в конце дыхательного цикла, предотвращая спадение ДП, формирование ателектазов и развитие вентиляционно-перфузионного несоответствия.
Описано четыре белка сурфактанта. Белки А и D участвуют в обеспечении защиты легких, тогда как сурфактантные белки В и С способствуют снижению поверхностного натяжения. АВСА3 является переносчиком пластинчатых телец органеллы, в которой хранится сурфактант в альвеолоцитах II типа, и играет важную роль в метаболизме фосфолипидов сурфактанта. Правильная экспрессия белков сурфактанта и АВСА3 зависит от ряда факторов транскрипции, в частности тиреоидного фактора транскрипции 1.
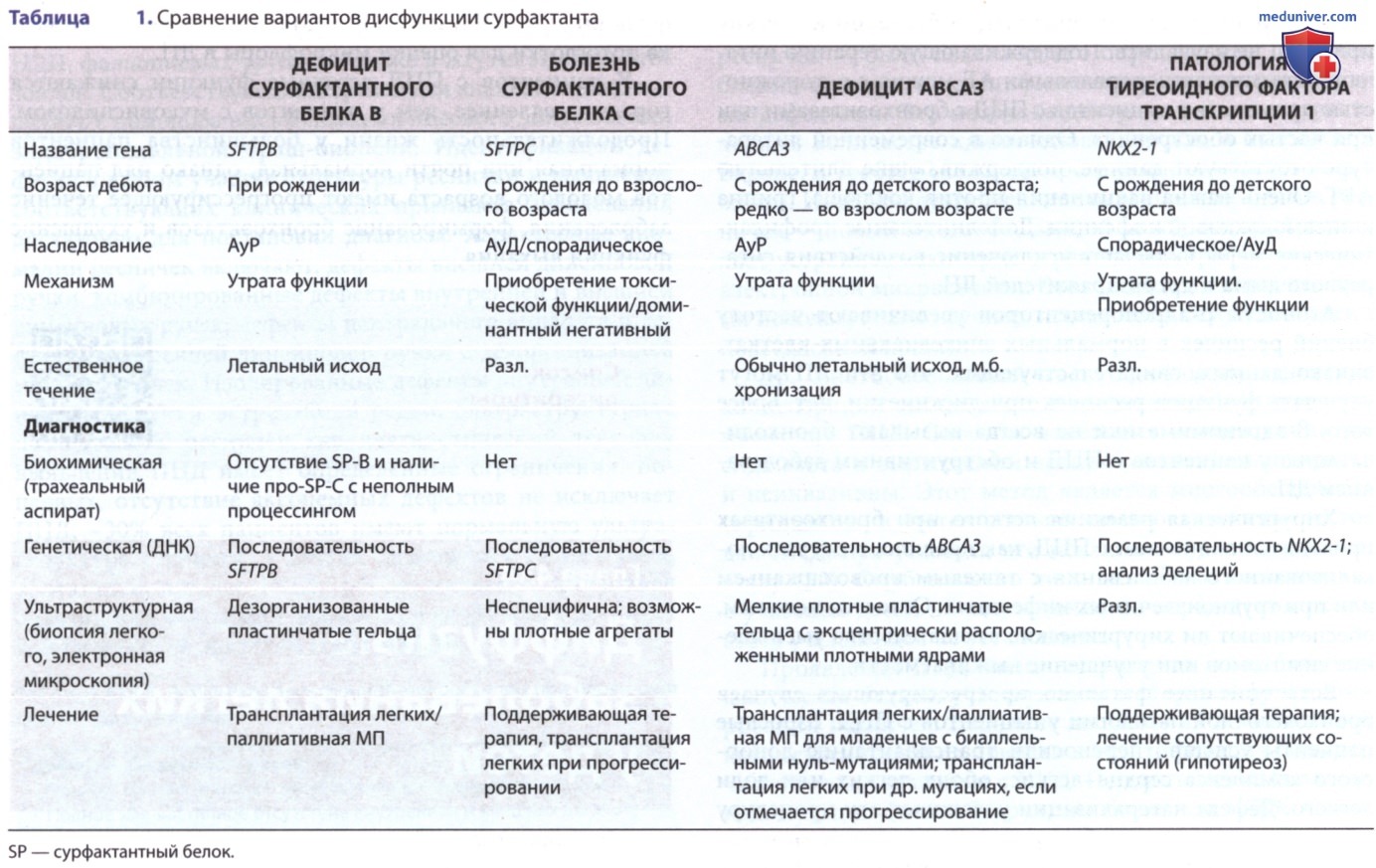
Два гена сурфактантного белка A (SFTPA1, SFTPA2) и один ген сурфактантного белка D (SFTPD) расположены на 10-й хромосоме, тогда как гены, кодирующие сурфактантный белок В (SFTPB), сурфактантный белок С (SFTPC), тиреоидный фактор транскрипции 1 (NKX2-1) и АВСА3, расположены на 2, 8, 14 и 16-й хромосомах человека соответственно. У человека идентифицированы наследственные нарушения сурфактантных белков В и С, АВСА3 и тиреоидного фактора транскрипции 1, которые в совокупности называются дисфункцией сурфактанта (табл. 1).
а) Патоморфология. Патоморфологически эти нарушения характеризуются уникальным сочетанием признаков, включая гиперплазию альвеолоцитов II типа, накопление альвеолярных макрофагов, утолщение и воспаление интерстициальной ткани, альвеолярный протеиноз. Исторически к этим нарушениям применяли ряд разл. описательных терминов, заимствованных из терапевтической практики (десквамативная интерстициальная пневмония, неспецифическая интерстициальная пневмония), а также заболеваний, присущих только младенческому возрасту (хронический пневмонит у детей).
Эти диагнозы убедительно указывают на дисфункцию сурфактанта, но не позволяют подтвердить генетическую природу заболевания. Поскольку прогноз и варианты наследования при этой патологии различаются в зависимости от генотипа, следует проводить генетическое тестирование.
1. Клинические проявления. У младенцев с наследственной недостаточностью сурфактантного белка В в раннем неонатальном периоде развивается ДН. Это АуР-заболевание клинически и рентгенологически похоже на РДС недоношенных детей, но обычно развивается у доношенных новорожденных. Начальная степень РДС м.б. разной, но заболевание прогрессирует, несмотря на проведение ИВЛ, заместительной терапии сурфактантом и введение ГКС. Дефицит сурфактантного белка В наблюдается у детей разных расовых и этнических групп. Почти все пациенты без трансплантации легких умирают в младенчестве, но при частичном дефиците возможна более длительная выживаемость.
При гетерозиготном носительстве мутации с утратой функции в гене SFTPB у взрослых курильщиков повышен риск развития обструктивного заболевания легких.
2. Генетические аспекты. Были идентифицированы многочисленные дефектные мутации в гене SFTPB. Самой распространенной является вставка двух пар оснований в кодон 133 (первоначально названная 121ins2, в настоящее время называемая c.397delCinsGAA, p.Prol33Glufs*95), которая приводит к сдвигу рамки считывания, нестабильной транскрипции сурфактантного белка В и отсутствию его производства. Эта мутация составляет 60-70% аллелей, обнаруженных на сегодняшний день у младенцев с дефицитом сурфактантного белка В, и присутствует у 0,07% европеоидов. Большинство др. мутаций выявляются в отдельных семьях. Описана также большая делеция, захватывающая два экзона гена сурфактантного белка В.
3. Диагностика. Быстрый и точный диагноз м.б. установлен с помощью анализа последовательности гена SFTPB, который доступен в клинических лабораториях. Разработано и внедрено в практику только секвенирование гена SFTPB, поскольку фенотип дефицита сурфактантного белка В совпадает с фенотипами др. вариантов дисфункции белков сурфактанта. Полигенные панели с использованием методов секвенирования нового поколения вытесняют использовавшееся ранее секвенирование отдельных генов. Для семей, в которых были идентифицированы мутации гена SFTPB, использование преимплантационной генетической диагностики/молекулярных анализов ДНК биоптатов ворсинок хориона/амниона позволяет заранее спланировать схему лечения.
Др. лабораторные тесты находятся в фазе разработки, включая анализ трахеального аспирата (промывных вод) на наличие/отсутствие сурфактантного белка В и на наличие пептидов — предшественников просурфактантного белка С с неполным процессингом, которые были обнаружены у младенцев с дефицитом сурфактантного белка В. Иммуноокрашивание биоптата легкого на сурфактантные белки также может подтвердить диагноз, хотя иммуногистохимические анализы на сурфактантные белки В и С в настоящее время используют только в научных исследованиях.
Окрашивание на сурфактантный белок В не проводят, но устойчивое внеклеточное окрашивание на просурфактантный белок С из-за его пептидов с неполным процессингом является диагностическим признаком дефицита сурфактантного белка В.
Такие исследования требуют выполнения биопсии легкого у тяжелобольного ребенка, но м.б. проведены на блоках легочной ткани, полученных во время вскрытия, что позволяет поставить диагноз ретроспективно. При электронной микроскопии отсутствие канальцевого миелина, дезорганизованные пластинчатые тельца и скопление патологических мультивезикулярных телец указывают на аномальную упаковку и секрецию липидов.
в) Аномалии гена сурфактантного белка С (нарушение метаболизма сурфактанта, легочное, 2 OMIM #610913). Сурфактантный белок С — это чрезвычайно гидрофобный белок с очень низкой молекулярной массой, который, наряду с сурфактантным белком В, усиливает св-ва снижения поверхностного натяжения фосфолипидов сурфактанта. Он является производным протеолитического процессинга более крупного белка-предшественника (просурфактантного белка С).
1. Клинические проявления. Клиническая картина у пациентов с мутациями SFTPC достаточно разнообразна. У некоторых пациентов уже при рождении отмечаются респираторные симптомы и рентгенологические данные, типичные для РДС. У др. людей заболевание манифестирует позже, от раннего младенческого до зрелого возраста, с постепенным развитием ДН, гипоксемии, задержкой физического развития и признаками интерстициального заболевания легких (ИЗЛ), что подтверждается рентгенологически.
У части пациентов заболевание манифестирует на пятом-шестом десятилетии жизни как легочный фиброз. Возраст дебюта и тяжесть заболевания различаются даже в семьях с одной и той же мутацией. Естественное течение также разнообразно: у некоторых пациентов состояние улучшается либо самостоятельно, либо на фоне терапии/длительной ИВЛ, у др. развивается стойкая ДН, а у ряда пациентов заболевание прогрессирует до необходимости проведения трансплантации легких. Эта вариабельность тяжести и течения заболевания, по-видимому, не коррелирует с конкретной мутацией и мешает точной оценке прогноза.
2. Генетические аспекты. Были идентифицированы многочисленные мутации в гене SFTPC у пациентов с острыми и хроническими заболеваниями легких от периода новорожденности до взрослого состояния. Мутации только в одном аллеле SFTPC достаточно, чтобы вызвать заболевание. Примерно половина этих мутаций возникают спонтанно, приводя к спорадическим заболеваниям, но остальные наследуются как доминантный признак. Замена изолейцина треонином в кодоне 73 (обозначаемая p.I73T/p.Ile73Thr) составляет 25-35% случаев, выявленных на сегодняшний день, но встречается редко (не идентифицирована в базе данных gnomAD у 123 000 человек).
Мутации гена SFTPC были обнаружены у представителей разл. расовых и этнических групп. Считается, что мутации в гене SFTPC приводят к образованию неправильно свернутого просурфактантного белка С, который накапливается в альвеолоцитах II типа и вызывает клеточное повреждение/изменяет нормальную в/клеточную маршрутизацию просурфактантного белка С.
3. Диагностика. Секвенирование SFTPC — единственный достоверный диагностический тест. Относительно небольшой размер гена облегчает проведение достаточно чувствительных анализов, но поскольку большинство мутаций гена SFTPC являются миссенс-мутациями, отличить истинные вызывающие заболевание мутации от редких, но функционально активных вариантов последовательности м.б. затруднительно. Иммуноокрашивание легочной ткани позволяет выявить агрегаты просурфактантного белка С, но только в научных исследованиях.
1. Клинические проявления. Заболевание легких, вызванное мутациями в гене АВСАЗ, обычно представляет собой тяжелую летальную форму, которая манифестирует сразу после рождения и клинически напоминает дефицит сурфактантного белка В/имеет хроническое течение, которое чаще всего проявляется на первом году жизни ИЗЛ, аналогично заболеванию, вызванному патологией сурфактантного белка С. Младенцы, которые являются гомозиготами/гетерозиготами по нулевым аллелям, что приводит к отсутствию экспрессии белка (т.е. нонсенс-мутации/мутации со сдвигом рамки считывания), как правило, погибают от ДН в неонатальном периоде, тогда как младенцы с др. типами мутаций имеют более вариабельный возраст дебюта заболевания и не всегда летальный исход.
Гетерозиготность по мутации АВСА3 может повышать риск развития РДС у недоношенных и доношенных детей, но в отличие от гетерозигот в конечном итоге заболевание легких у них может полностью разрешиться.
2. Генетические аспекты. Рецессивные мутации в АВСА3 были впервые описаны у новорожденных, погибших от РДС, но теперь они идентифицированы и у младенцев более старшего возраста и детей с ИЗЛ. Отмечается значительная аллельная гетерогенность: идентифицированы >400 мутаций, разбросанных по всему гену, большинство из которых являются семейно-специфическими. Наличие нулевых мутаций на обоих аллелях, что, как предполагается, исключает синтез АВСАЗ, приводит к раннему началу заболевания и неизменно летальному исходу.
Миссенс-мутация, которая приводит к замене валина на глутамин в кодоне 292 (p.E292V/p.Glu292Val) в сочетании со второй мутацией АВСА3, была обнаружена у детей с тяжелой неонатальной ДН и у детей старшего возраста с ИЗЛ. Она встречается с частотой 0,7% у европеоидов. Мутации АВСА3 были идентифицированы у представителей разл. расовых и этнических групп. Точная частота заболевания не известна, но крупномасштабные проекты по секвенированию показывают, что общая частота носителей мутации АВСА3 может составлять 1:50-70 человек. Т.о., дефицит АВСА3 может способствовать значительной части необъяснимых летальных заболеваний легких у доношенных детей и ИЗЛ у детей старшего возраста.
4. Диагностика. Анализ последовательности АВСА3 доступен в клинических лабораториях и является самым точным методом диагностики. Значительные различия в АВСА3 требуют внимательной интерпретации в отношении функциональности отдельного варианта и его вклада в клиническую картину. Кроме того, анализ последовательности не является 100% чувствительным, т.к. возможны функционально значимые мутации в нетранслируемых областях, которые обычно не анализируются. В этих ситуациях биопсия легкого с электронной микроскопией для изучения морфологии пластинчатых телец м.б. полезным дополнением при диагностике заболевания.
При мутациях АВСА3 обнаруживают мелкие пластинчатые тельца, содержащие электронно-плотные включения. Эти данные подтверждают гипотезу о том, что функция АВСАЗ необходима для биогенеза пластинчатых телец. Биохимические маркеры для постановки диагноза отсутствуют.
д) Заболевание, вызванное мутациями в NKX2-1 (тиреоидный фактор транскрипции 1, хореоатетоз, гипотиреоз и нарушения дыхания у новорожденных, OMIM #600635):
1. Клинические проявления. Большая делеция области хромосомы 14 (14q13.3), включающей локус NKX2-1, была впервые обнаружена у младенца с гипотиреозом и РДС новорожденных. С тех пор у людей с гипотиреозом, легочными заболеваниями и неврологическими симптомами, включая доброкачественную семейную хорею, были описаны многочисленные большие делеции с участием локуса NKX2-1 и сцепленных генов, включая миссенс-мутации, мутации со сдвигом рамки считывания, нонсенс-мутации и небольшие инсерции/делеции, разбросанные по всему гену. Дисфункцию всех трех систем органов называют синдромом «мозг-легкие-ЩЖ», но заболевание также может проявляться поражением только в одной/двух системах органов.
Заболевание легких м.б. разл. выраженности, от тяжелого и в конечном итоге летального РДС новорожденных до хронического заболевания легких как у детей, так и у взрослых.
Описаны рецидивирующие легочные инфекции, вероятно, вызванные снижением экспрессии легочных коллектинов, сурфактантных белков А и D, но также, возможно, в результате уменьшения экспрессии др. белков. Четких корреляций между генотипом и фенотипом не обнаружено, но дети с полными делециями генов, как правило, имеют более тяжелое заболевание с ранним дебютом. Это также м.б. связано с делецией др. сцепленных генов. Клинические данные ограниченны, но легочный фенотип зависит от экспрессии наиболее затронутых генов-мишеней NKX2-1. У детей со сниженной экспрессией сурфактантного белка В/АВСА3 возможно развитие ОДН в неонатальном периоде, тогда как дети со сниженной экспрессией сурфакантного белка С/легочного коллектина с большей вероятностью будут иметь хроническое течение заболевание легких.
2. Генетические аспекты. Ген NKX2-1 небольшой, содержит <3000 оснований и всего три экзона. Тиреоидный фактор транскрипции 1 экспрессируется не только в легких, ЩЖ и ЦНС. В легких он важен для экспрессии широкого спектра белков, включая белки сурфактанта, АВСА3, секреторный белок клеток Клара (Clara Мах) и многие другие. Были обнаружены два транскрипта, которые различаются в зависимости от того, находится ли сайт начала транскрипции в первом/втором экзоне, но более короткий транскрипт преобладает в легких. Считается, что большинство мутаций приводит к утрате функции, причем механизмом заболевания является гаплонедостаточность, но описаны дискордантные влияния на разл. гены-мишени. Мутации с утратой функции в NKX2-1 редки, и распространенность болезни не известна.
Были обнаружены мутации у представителей разл. этнических групп. Большинство зарегистрированных мутаций появляются de novo, что приводит к спорадическим случаям заболевания, но также описаны и семейные случаи заболевания, передающееся как доминантный признак.
3. Диагностика. Анализ последовательности гена NKX2-1 возможен в клинических лабораториях и является основным методом диагностики. Поскольку делеции составляют значительную часть описанных мутантных аллелей, также следует применять сравнительный анализ геномной гибридизации, анализ мультиплексной амплификации лигированных зондов и использование методологии секвенирования нового поколения. Мутации в одном аллеле достаточно, чтобы вызвать заболевание. Описаны случаи изолированного легочного поражения, но у большинства зарегистрированных пациентов отмечают изменения в >2 органах. Т.о., наличие гипотиреоза/неврологической патологии у пробанда, семейный анамнез хореи требуют исключения данного заболевания.
Самым специфическим неврологическим признаком является хорея, но также возможна гипотония, задержка развития, атаксия и дизартрия. У младенцев мышечная слабость, гипотония м.б. обусловлены тяжестью бронхолегочной патологии/сопутствующим гипотиреозом. У пациентов гипотиреоз м.б. компенсированным с пограничным снижением уровня Т4 (тироксина) и высокими уровнями ТТГ. Патология легких, вызванная мутациями NKX2-1, неспецифична и может напоминать др. виды дисфункции сурфактанта. Для дисфункции гена NKX2-1, который важен для развития легких, характерны аномалии роста и развития легких. Исследования с иммуноокрашиванием белков сурфактанта показали противоречивые результаты. Характерных изменений при электронной микроскопии также получено не было.
е) Лечение дисфункции сурфактанта. Практически все пациенты с дефицитом сурфактантного белка В умирают в первый год жизни. Стандартные вмешательства в ОРИТ новорожденных позволяют поддерживать функцию органов и систем в течение ограниченного времени (от нескольких недель до нескольких месяцев). Заместительная терапия сурфактантами неэффективна. Трансплантация легких оказалась успешной, но предтрансплантационная, трансплантационная и посттрансплантационная терапевтическая и хирургическая МП является узкоспециализированной и доступна только в педиатрических центрах трансплантации легких; быстрая диагностика имеет решающее значение, если должен решаться вопрос о пересадке легких. В этой группе больных актуально оказание паллиативной МП.
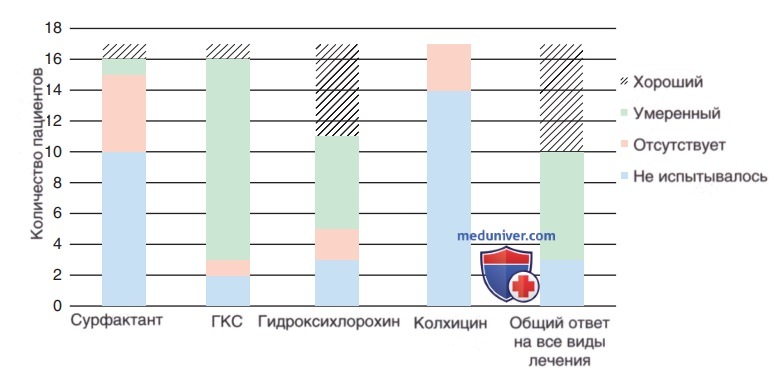
Специфического лечения при патологии легких, вызванных мутациями в SFTPC/АВСА3, нет. В лечении используют ГКС, хинолоны и макролиды, но оценка их эффективности и безопасности не проводилась (рис. ниже). Младенцы с тяжелой и прогрессирующей ДН, вызванной дефицитом АВСАЗ, м.б. кандидатами на трансплантацию легких. Разл. течение болезни у пациентов с мутациями SFTPC и детей старшего возраста с дефицитом АВСА3 затрудняет прогнозирование исхода. Независимо от диагноза пациентам с прогрессирующей и рефрактерной ДН проводят трансплантацию легких.
Ответ на терапию у 17 пациентов с мутациями гена сурфактантного белка С.
Лечение пациентов с мутациями NKX2-1 в основном является симптоматическим. Гипотиреоз следует лечить с помощью заместительной терапии гормонами ЩЖ, ГКС и др. ЛП, используемые при иных типах дисфункции сурфактанта, не проходили формальную оценку. При прогрессирующем заболевании легких показана трансплантация. Разл. степень прогрессирования заболевания и наличие внелегочных поражений могут особенно затруднить оценку и выбор пациентов для трансплантации.
Родителям детей с дисфункцией сурфактанта и их ближайшим родственникам следует предложить генетическое консультирование и пренатальную диагностику, чтобы избежать рождения больного ребенка при будущих беременностях.