Синдромы наследственной периодической лихорадки — группа моногенных заболеваний, которые проявляются повторяющимися приступами лихорадки с сопутствующим воспалением плевры и/или брюшины, артритом и кожной сыпью разл. типа. Определенные расстройства проявляются повторяющимися эпизодами воспаления, которое не всегда сопровождается лихорадкой. Поэтому термин «системные аутовоспалительные заболевания» используется с целью охватить все заболевания, которые проявляются, по всей видимости, ничем не провоцируемыми эпизодами воспаления, без высокого титра ауто-АТл/АГн-специфических Т-клеток, обычно наблюдаемых при аутоиммунных заболеваниях.
В то время как аутоиммунные заболевания возникают вследствие нарушения адаптационной иммунной системы и вызываются клетками — В-/Т-эффекторными лимфоцитами, — аутовоспалительные заболевания в значительной степени представляют собой нарушения филогенетически более примитивной врожденной иммунной системы и опосредуются миелоидными эффекторными клетками и рецепторами, кодируемыми множественными эмбриональными генами. При аутовоспалительных заболеваниях наблюдается эпизодическое/стойкое воспаление, характеризующееся острофазным ответом с повышением СОЭ, СРВ и амилоида А (АА) сыворотки. У некоторых пациентов нелеченные аутовоспалительные заболевания со временем приводят к развитию АА-амилоидоза.
Важно заметить, что аутовоспалительные заболевания встречаются редко, в то время как лихорадки, обусловленные др. патологией, в детском возрасте очень распространены. Подход к ребенку с лихорадкой должен включать подробный сбор анамнеза, физикальное обследование и лабораторные исследования в разумных пределах с целью исключить др. состояния, которые м.б. причиной лихорадки, включая аутоиммунные заболевания и ЗНО (табл. 1). Если в анамнезе есть свидетельства о рецидивирующих инфекциях, сопровождавшихся лихорадкой, следует рассмотреть иммунодефицит как один из возможных вариантов.
Если результаты осмотра обнадеживают, эпизоды воспаления разрешаются, и в остальном ребенок чувствует себя хорошо, у него нет необычных симптомов, то в таких случаях показано только динамическое наблюдение, поскольку эти эпизоды, вероятно, прекратятся полностью сами собой по мере созревания иммунной системы ребенка.
а) Классификация аутовоспалительных заболеваний. Из-за быстро растущего числа аутовоспалительных заболеваний и разнообразия их клинических проявлений трудно сгруппировать эти расстройства. Некоторые аутовоспалительные заболевания проявляются выраженной лихорадкой и известны как синдромы наследственной периодической лихорадки. В эту группу входят 2 заболевания с АуР-наследования, семейная средиземноморская лихорадка (FMF; англ, familial Mediterranean fever; MIM249100) и гипериммуноглобулинемия D с синдромом периодической лихорадки (HIDS; англ. hyper-IgD syndrome; MIM260920).
Синдромы наследственной периодической лихорадки с АуД-типом наследования включают периодический синдром, связанный с рецептором ФНО (TRAPS; англ. Tumor necrosis factor Receptor-Associated Periodic Syndrome; MIM191190), и спектр расстройств, известных как криопирин-ассоциированные периодические синдромы (CAPS; англ. cryopyrin-associated periodic syndromes)/криопиринопатии. Криопиринопатии включают семейный холодовой аутовоспалительный синдром (FCAS1; англ. familial cold autoinflammatory syndrome; MIM120100), синдром Макла-Уэллса (MWS; англ. Muckle-Wells syndrome; MIM191100) и мультисистемное воспалительное заболевание с неонатальным началом (NOMID; англ. Neonatal-Onset Multisystem Inflammatory Disease; MIM607115) (также известное как хронический детский неврологический, кожный и суставной синдром, CINCA) (табл. 2).
Разнообразные менделевские аутовоспалительные заболевания могут проявляться/не проявляться выраженной лихорадкой, и поэтому не учитываются в числе синдромов периодической лихорадки, но для них характерны непрерывные/повторяющиеся эпизоды спонтанного воспаления с уникальными клиническими характеристиками. К ним относятся: синдром пиогенного артрита с гангренозной пиодермией и акне (PAPA; англ. Pyogenic Artritis, Pyoderma gangrenosum, Acne; MIM604416), дефицит антагониста рецептора IL-1 (DIRA; англ. deficiency of the interleukin-1 receptor antagonist; MIM612852), синдром Блау, вызванный мутациями в гене N0D2 (также известный как саркоидоз с ранним началом; MIM186580), аутовоспаление с дефицитом АТл, ассоциированных с фосфолипазой Сγ2, и иммунной дисрегуляцией (APLAID; англ, autoinflammation with phospholipase Cγ2-associated antibody deficiency and immune dysregulation; MIM614878), а также дефицит аденозиндезаминазы 2-го типа (DADA2; англ, deficiency of adenosine deaminase-2).
Прочие расстройства включают: врожденную сидеробластную анемию с В-клеточным иммунодефицитом, периодической лихорадкой и задержкой развития (SIFD; англ. sideroblastic anemia with В-cell immunodeficiency, periodic fevers, and developmental delay), которая вызывается биаллельными мутациями гена TRNT1 (MIM616084), аутовоспаление с детским энтероколитом, вызванное мутациями в NLRC4 (AIFEC; англ. autoinflammation with infantile enterocolitis caused by mutations in NLRC4; MIM616060), FCAS2, CARD14 (MIM607211), и дефицит антагониста рецептора IL-36 (DITRA; англ. deficiency in IL-36 receptor antagonist; 614204).
В дополнение к ранее перечисленным аутовоспалительным заболеваниям, выделяют группу заболеваний, называемых интерферонопатиями, и характеризующихся избыточной экспрессией IFN. IFN 1-го типа (напр., IFN-α, IFN-β) — это цитокины, экспрессируемые многими клетками в ответ на вирусные инфекции. Заболевания, которые приводят к спонтанной продукции IFN и клинике воспаления, включают STING-ассоциированную васкулопатию младенческого возраста (SAVI; англ. STING-associated vasculopathy of infancy; MIM615934) и хронический атипичный нейтрофильный дерматоз с липодистрофией и повышенной ТТ (CANDLE; англ. chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; MIM256040).
Также существует ряд аутовоспалительных заболеваний со сложным типом наследования. К ним относятся PFAPA и хронический рецидивирующий мультифокальный остеомиелит (CRMO; англ. chronic recurrent multifocal osteomyelitis; MIM259680). Прочие генетически сложные заболевания, которые иногда относят к аутовоспалительным, включают ЮИА с системным началом, болезнь Бехчета и болезнь Крона.
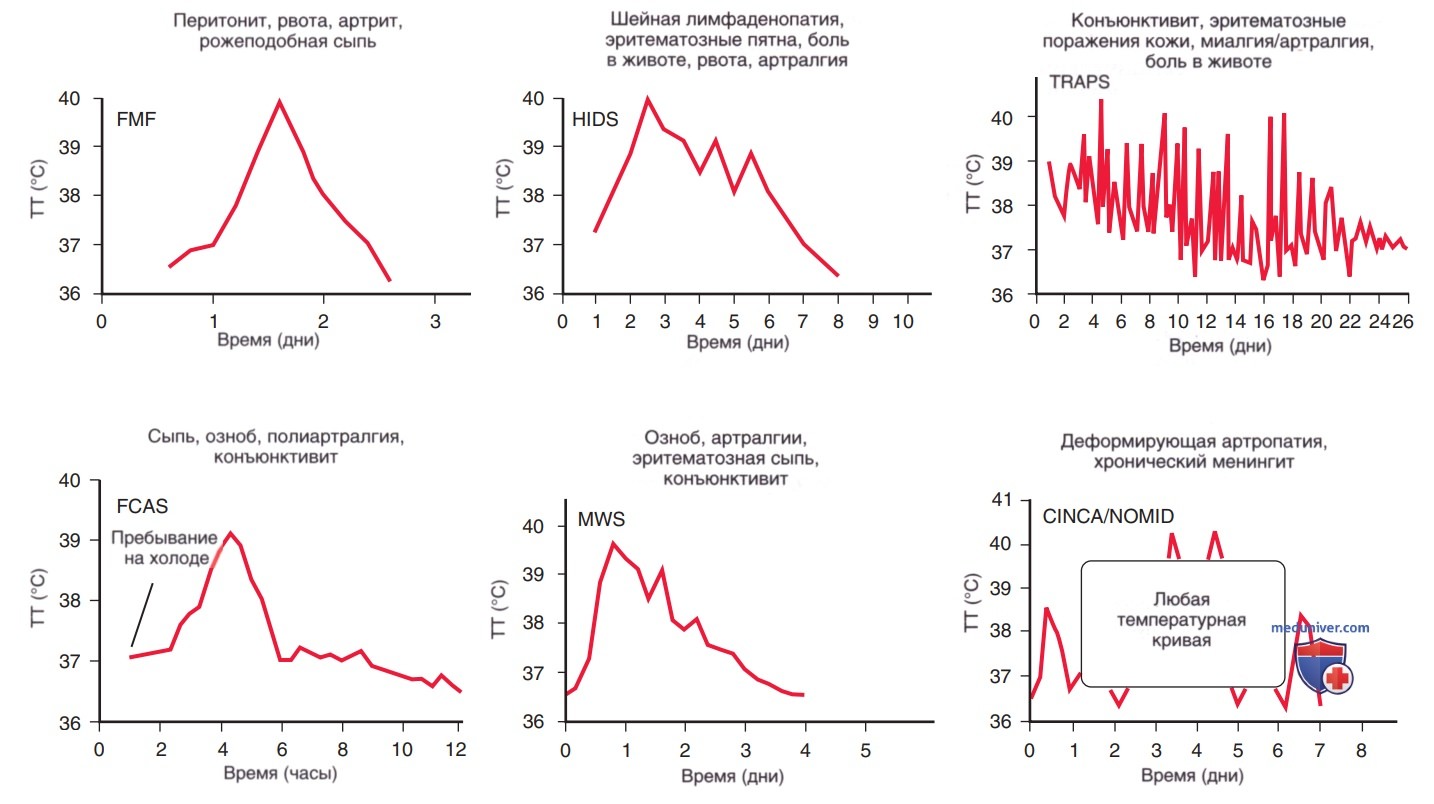
Порой бывает сложно отличать аутовоспалительные заболевания друг от друга вследствие вариабельности их проявлений и большого количества сходных черт. Некоторые из них имеют характерные паттерны лихорадки (рис. 1), тогда как для других характерны определенные кожные признаки, которые могут помочь в диагностике лихорадки (табл. 3). Некоторые из них могут иметь характерные физикальные особенности/поражать определенные органы. Часть этих заболеваний сопровождается поражением костей (табл. 4). М.б. полезны и др. клинические признаки, напр., этническая принадлежность пациента, возраст дебюта, триггеры, результаты лабораторных исследований и ответ на терапию (табл. 5). В настоящее время все чаще стали использовать генетические панели для скрининга большинства, если не всех этих дефектов, в рамках одного теста, вместо индивидуального генетического тестирования, основанного на клинических данных.
Рисунок 1. Характерные паттерны изменения температуры тела во время воспалительных атак при семейных аутовоспалительных синдромах. Для каждого синдрома наблюдается значительная вариабельность, и даже у отдельных пациентов характер лихорадки может сильно различаться от эпизода к эпизоду. Обратите внимание на разность шкал времени по оси х. FMF — семейная средиземноморская лихорадка; CINCA/NOMID — хронический детский неврологический кожный и суставной синдром/мультисистемное воспалительное заболевание с неонатальным началом; FCAS — семейный холодовой аутовоспалительный синдром; HIDS — синдром гипериммуноглобулинемии D; MWS — синдром Макла-Уэллса; TRAPS — периодический синдром, связанный с рецептором ФНО
б) Аутовоспалительные заболевания с периодической или выраженной лихорадкой. Первые описания аутовоспалительных расстройств заостряли внимание на генетических заболеваниях, которые проявлялись выраженной лихорадкой и синдромами периодической лихорадки. Однако по мере открытия новых аутовоспалительных расстройств стало ясно, что многие воспалительные заболевания могут протекать и без лихорадки.
1. Семейная средиземноморская лихорадка. FMF — это АуР-наследуемое аутовоспалительное заболевание, обычно характеризующееся рецидивирующими, непродолжительными (1-3 дня), самопроходящими эпизодами лихорадки, серозитом, моно-/олигоартритом и рожеподобной сыпью, иногда осложняющееся АА-амилоидозом. У большинства пациентов с FMF симптомы проявляются еще в детстве, у 90% из них — раньше 20 лет. Клинические признаки могут включать: лихорадку, серозит, проявляющийся болью в груди (плеврит)/сильной абдоминальной болью (перитонит), артритом и сыпью. Плевральная боль обычно односторонняя, тогда как боль в животе (стерильный перитонит) м.б. генерализованной/локализоваться в пределах 1 квадранта, как и при др. формах перитонита. FMF-ассоциированный артрит поражает, в основном, крупные суставы, может сопровождаться обильным нейтрофильным выпотом, обычно бывает неэрозивным и не приводит к деструктивным изменениям.
Патогномоничное поражение кожи — рожеподобная эритематозная сыпь, которая чаще локализуется в области голеностопного сустава/тыльной поверхности стопы (рис. 2). Прочие клинические признаки включают: боль в мошонке, обусловленную воспалением влагалищной оболочки яичка, фебрильную миалгию, миалгию, вызванную ФН (особенно у детей), а также разл. формы васкулита, включая пурпуру Шенляйна-Геноха(Schonlein, Henoch) у 5% педиатрических пациентов. Эпизоды FMF могут провоцироваться стрессом, менструацией и инфекциями. Между обострениями у пациентов, как правило, отсутствуют симптомы, но маркеры воспаления м.б. постоянно повышены. Частота приступов может варьировать от еженедельных до 1-2 в год. В табл. 6 перечислены диагностические критерии FMF.
Рисунок 2. Характерная рожеподобная эритема при семейной средиземноморской лихорадке. Такая сыпь появляется во время обострения и затрагивает голеностопный сустав/тыльную сторону стопы
Развитие FMF обусловлено АуР-мутациями в MEFV, гене, кодирующем белок, состоящий из 781 аминокислоты и обозначаемый как пирин (греч. «лихорадка»). Пирин экспрессируется в гранулоцитах, моноцитах и дендритных клетках, а также в перитонеальных, синовиальных и дермальных фибробластах. Ок. 90 аминокислот, находящихся на N-конце пирина, являются прототипом мотива (домен PYRIN), который опосредует межбелковые взаимодействия и обнаруживается в >20 разл. белках человека, которые регулируют воспаление и апоптоз. Многие из FMF-ассоциированных мутаций пирина обнаруживаются в С-концевом В30.2 домене пирина, кодируемом экзоном 10-го гена MEFV.
Более 50 таких мутаций перечислены в онлайн-базе данных, и практически все они представляют собой миссенс-замены. Гомозиготность по мутации M694V может вызывать более раннее начало заболевания, развитие артрита и повышать риск амилоидоза. Замена глутамина на глутаминовую кислоту в остатке 148 (E148Q) считается легкой мутацией/функциональным полиморфизмом пирина. Распространенность носительства FMF-мутаций среди некоторых средиземноморских популяций очень высока, что предполагает возможность клинических проявлений у гетерозигот.
FMF встречается в основном среди этнических групп средиземноморского происхождения, чаще всего среди евреев, турок, армян, арабов и итальянцев. Из-за более высокой частоты мутации M694V гена MEFV FMF тяжелее протекает и легче распознается у евреев-сефардов (Северная Африка), чем у евреев-ашкенази (Восточная Европа). С появлением генетического тестирования FMF, доказанная генетически, была зарегистрирована во всем мире, хотя и с меньшей частотой, чем в Средиземноморском бассейне и на Ближнем Востоке.
P.S. FMF (англ. Familial Mediterranean Fever) — семейная средиземноморская лихорадка.
Посредством взаимодействия с PYRIN-доменом пирин может активировать каспазу-1 — фермент, который превращает молекулу про-IL-1β массой 31 кДа в биологически активный IL-1β массой 17 кДа, который является основным медиатором лихорадки и воспаления. Мутации, вызывающие FMF, ведут к усилению активации каспазы-1 и IL-1β-зависимого воспаления с эффектом дозы гена. Этим может объясняться тот факт, что до 30% гетерозиготных носителей FMF-мутаций имеют биохим. признаки воспаления.
Ежедневный профилактический прием колхицина снижает частоту, продолжительность и интенсивность приступов FMF. Этот режим терапии также предотвращает развитие системного АА-амилоидоза. Колхицин, как правило, хорошо переносится и безопасен для детей, наиболее частыми побочными эффектами являются диарея и др. жалобы со стороны ЖКТ. У некоторых пациентов на фоне приема колхицина развивается непереносимость лактозы. Побочные эффекты со стороны ЖКТ можно свести к минимуму, если начать терапию с низкой дозы (для детей младшего возраста 0,3 мг/сут), постепенно ее повышая. Также может наблюдаться дозозависимое повышение трансаминаз. Угнетение костно-мозгового кроветворения редко наблюдается при дозах, применяемых при FMF.
Детям могут потребоваться дозы колхицина, аналогичные дозам для взрослых (1-2 мг/сут), что свидетельствует о более быстрой метаболизации ЛП у детей, чем у взрослых. Не всегда можно найти хорошо переносимую дозу колхицина, при которой удается добиться исчезновения всех симптомов, но у 90% пациентов наблюдается заметное уменьшение симптомов заболевания. Небольшой процент пациентов с FMF не реагирует на терапевтические дозы колхицина/не переносит их. Для колхицин-резистентных пациентов >2 лет одобрено применение канакинумаба, эффективность которого доказана в многоцентровом РКИ. Исследование, основанное на роли пирина в активации IL-1β, продемонстрировало безопасность и эффективность рилрнацепта, ингибитора IL-1, при FMF. Имеются сообщения об эффективности анакинры — рекомбинантного антагониста рецептора IL-1R.
Амилоидоз — наиболее серьезное осложнение FMF, и в его отсутствие продолжительность жизни пациентов не отличается от средней в популяции. Амилоидоз может развиться, когда белок АА, реактант острой фазы, обнаруживаемый в крови в чрезвычайно высоких концентрациях во время приступов FMF, расщепляется с образованием 76-аминокислотного фрагмента, который неправильно складывается и эктопически депонируется, чаще всего в почках, ЖКТ, селезенке, легких, яичках, ЩЖ и надпочечниках. Редко встречается амилоидоз сердца. Макроглоссия и амилоидная невропатия обычно не наблюдаются при амилоидозе на фоне FMF. Наиболее частый симптом АА-амилоидоза — протеинурия. Диагноз обычно подтверждается биопсией прямой кишки/почек.
Существует небольшое количество сообщений о случаях, в основном из стран Ближнего Востока, при которых амилоидоз может даже предшествовать явным приступам FMF, предположительно вследствие субклинического воспаления. Факторы риска развития амилоидоза при FMF: гомозиготность по мутации M694V MEFV, полиморфизм сывороточного гена АА (кодирующего АА), несоблюдение режима лечения колхицином, мужской пол и семейный анамнез АА-амилоидоза. По неясным причинам страна происхождения также является важным фактором риска развития амилоидоза при FMF. Пациенты, выросшие на Ближнем Востоке, имеют гораздо более высокий риск его развития, чем генотипически идентичные пациенты, выросшие на Западе. Агрессивное пожизненное подавление реактантов острой фазы — цель терапии у пациентов с амилоидозом и FMF, зарегистрированные случаи показывают, что приверженность этой тактике может привести даже к резорбции имеющихся отложений амилоида.
При отсутствии лечения амилоидоз на фоне FMF ведет к неумолимому прогрессированию почечной недостаточности, часто в течение 3-5 лет.
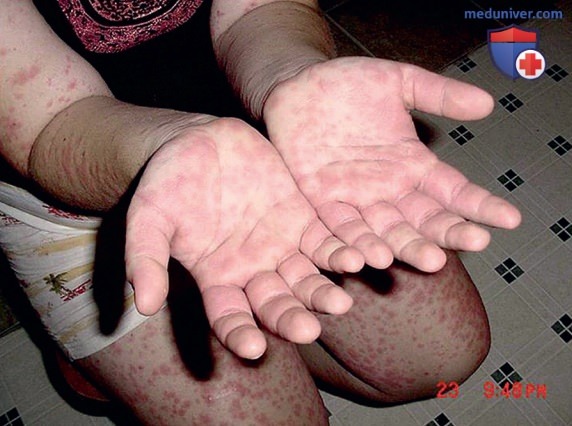
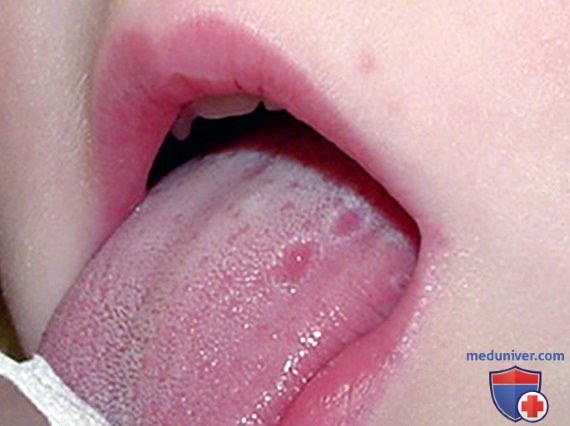
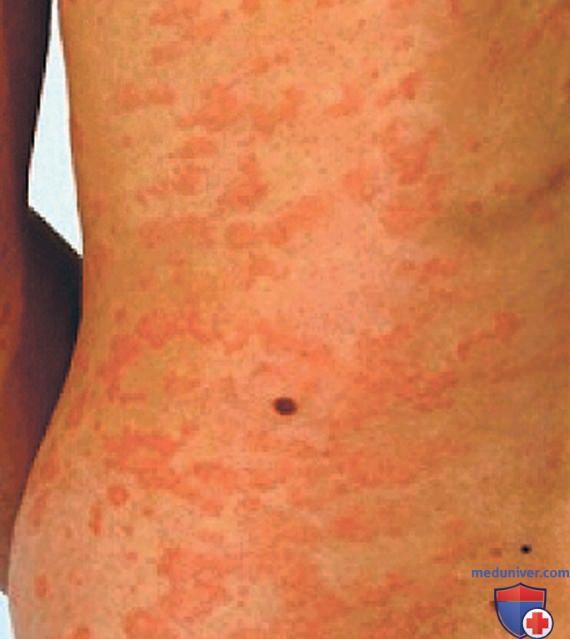
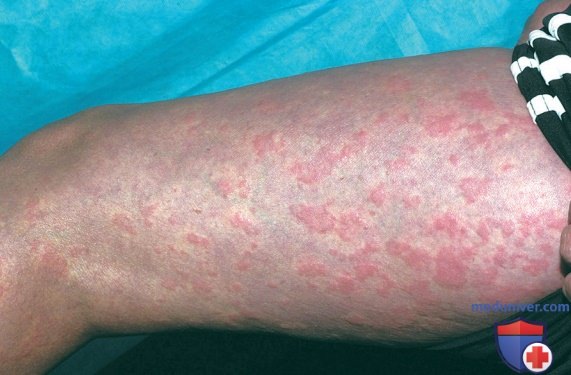
2. Гипериммуноглобулинемия D с синдромом периодической лихорадки. HIDS, также известная как дефицит мевалонаткиназы, была первоначально описана в когорте голландских пациентов и встречается в основном у пациентов североевропейского происхождения. HIDS наследуется по АуР-типу и вызывается мутациями гена MVK, кодирующего мевалонаткиназу. Клинические признаки HIDS обычно проявляются в первые 6 мес жизни. Приступы лихорадки длятся 3-7 дней и сопровождаются болями в животе, часто с диареей, тошнотой и рвотой. Др. клинические проявления включают шейную лимфаденопатию, диффузную макулярную сыпь, афтозные язвы, головные боли и периодическую спленомегалию (рис. 3-5). Артрит/артралгия м.б. олигоартикулярными/полиартикулярными.
Рисунок 3. Полиморфная сыпь на кистях, руках и ногах пациента с синдромом гипер-IgD
Рисунок 4. Петехии на ноге пациента с синдромом гипер-IgD во время приступа лихорадки
Рисунок 5. Афтозное изъязвление языка у пациента с синдромом гипер-IgD
Сообщалось также о случаях воспалительно-подобных заболеваний и Кавасаки (Kawasaki)-подобных случаях. Приступы часто провоцируются интеркуррентными заболеваниями, вакцинацией и хирургическими вмешательствами. В семьях часто случаются вспышки во время дней рождения, каникул и семейных праздников. Симптомы HIDS могут сохраняться годами, но, как правило, у взрослых становятся менее выраженными. Пациенты с HIDS обычно живут столько же, сколько средний человек в популяции. В отличие от FMF и TRAPS, АА-амилоидоз в этих случаях встречается довольно редко. Полный дефицит мевалонаткиназы приводит к мевалоновой ацидурии, которая проявляется тяжелой умственной отсталостью, атаксией, миопатией, катарактой и задержкой развития.
Мевалонаткиназа экспрессируется во многих тканях и катализирует превращение мевалоновой кислоты в 5-фосфомевалоновую кислоту при биосинтезе изопреноидов ХС и нестерола. У пациентов с HIDS-ассоциированными мутациями ферментативная активность мевалонаткиназы значительно снижена, но не отсутствует полностью. Пациенты с HIDS обычно имеют низкий уровень ХС в сыворотке крови, но дефицит изопреноидов может вызвать повышение продукции IL-1β за счет аберрантной активации минорной гуанозинтрифосфатазы Rael. Повышение ТТ способно еще больше усугублять этот процесс за счет более полного ингибирования активности мевалонатки-назы, что может приводить к формированию петли обратной связи.
Диагноз HIDS м.б. подтвержден обнаружением 2 мутаций в MVK (у 10% пациентов с типичной, на первый взгляд, картиной заболевания обнаруживается только 1 идентифицируемая мутация)/обнаружением повышенного уровня мевалоната в моче во время приступов. HIDS-мутации распределены по всему белку мевалонаткиназы, но 2 наиболее распространенные — это замена изолейцина на валин в остатке 377 (V377I), вариант, который наиболее распространен в голландской популяции, и замена треонина на изолейцин в остатке 268 (I268T). Одновременное повышение сывороточных уровней IgD наблюдается не всегда, особенно у маленьких детей.
Также м.б. повышен уровень IgA. И наоборот, уровни IgD в сыворотке могут повышаться и при др. аутовоспалительных заболеваниях и некоторых хронических инфекциях. Во время приступов часто наблюдается лейкоцитоз и повышение уровней реактантов острой фазы и провоспалительных цитокинов в сыворотке крови. В табл. 7 перечислены ДК HIDS.
P.S. HIDS — синдром гипериммуноглобулинемии D (Hyper-IgD Syndrome).
Стандарты лечения HIDS все еще разрабатываются. Небольшая доля пациентов отвечают на колхицин, а более легкий вариант заболевания может отвечать на НПВП. Применение ГКС ограничено. Обнадеживают результаты небольших испытаний этанерцепта и периодического/ежедневного применения анакинры при HIDS. Для пациентов с HIDS >2 лет одобрено применение канакинумаба, эффективность которого доказана в многоцентровом РКИ*.
P.S. * Канакинумаб в лечении периодической болезни и др. аутовоспалительных заболеваний — исследование CLUSTER, журнал «Клиническая фармакология и терапия», выпуск 2018.4.
3. Периодический синдром, ассоциированный с рецептором фактора некроза опухоли. TRAPS характеризуется рецидивирующими лихорадками и локализованным воспалением и наследуется по АуД-типу. TRAPS имеет ряд отличительных клинических и иммунологических особенностей. Он был впервые обнаружен у пациентов ирландского происхождения и получил название семейной ирландской лихорадки с целью отличия от FME Текущая номенклатура была предложена в связи с тем, что мутации TNFRSF1A были обнаружены не только в исходной ирландской семье, но и в семьях из др. этнических групп. TNFRSF1A кодирует рецептор ФНО-α массой 55 кДа (обозначенный р55, TNFRl/CD120a), который широко экспрессируется на ряде типов клеток. Второй рецептор, массой 75 кДа, в значительной степени ограничен лейкоцитами.
Пациенты с TRAPS обычно обращаются к врачу в первое десятилетие жизни по поводу обострений, которые возникают с переменной частотой, и часто имеют значительно большую длительность, чем обострения FMF/HIDS. Эпизоды лихорадки при TRAPS длятся не <3 дней, но могут сохраняться и в течение нескольких недель. При этом может развиваться поражение плевры и брюшины. Иногда пациенты обращаются с клиникой острого живота, при обследовании у таких больных обнаруживается стерильный перитонит, иногда со спайками после предыдущих эпизодов. У пациентов может наблюдаться тошнота и запор на начальном этапе обострения, который к окончанию эпизода переходит в диарею. Поражения глаз включают периорбитальный отек и конъюнктивит. Пациенты с TRAPS могут также страдать от тяжелой миалгии, а при визуализации определяются очаги отека в отдельных группах мышц.
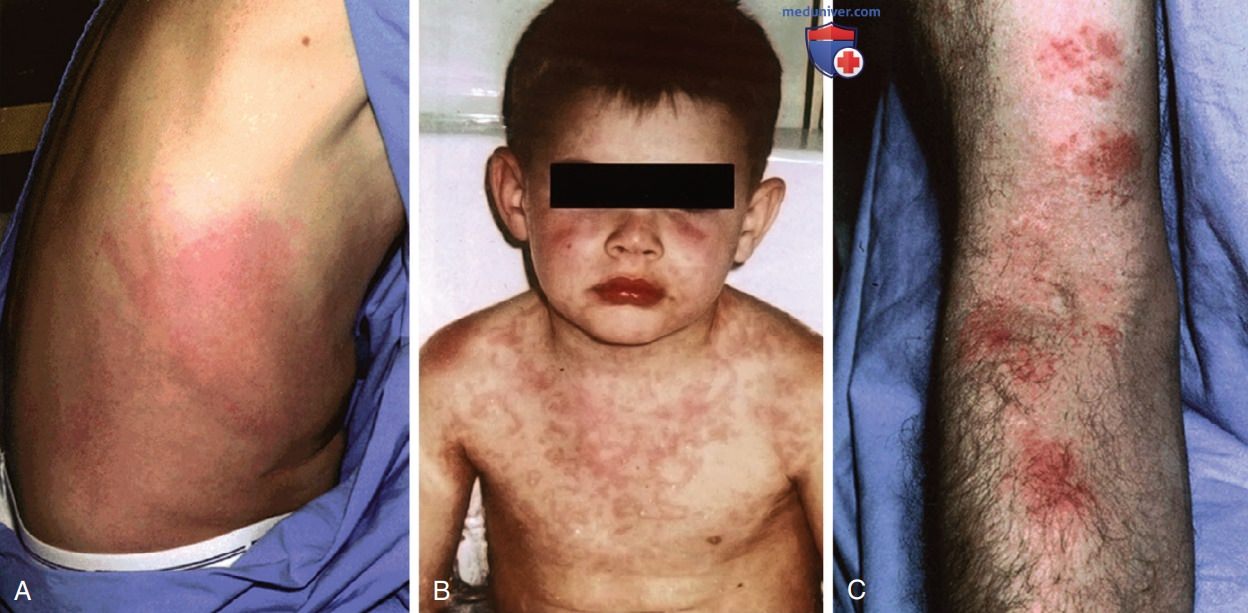
У пациентов с TRAPS отмечаются разнообразные высыпания, но наиболее распространена эритематозная макулярная сыпь, при биопсии которой обнаруживаются поверхностные и глубокие периваскулярные инфильтраты мононуклеарных клеток. Пациенты часто сообщают о миграции сыпи и миалгии по конечности в дистальном направлении, что может напоминать целлюлит. Кроме того, встречаются эритематозные кольцевидные пятна и серпигинозная сыпь (рис. 6). У 10-15% пациентов с TRAPS может развиться АА-амилоидоз, факторами риска развития которого являются: наличие мутаций цистеина и семейный анамнез амилоидоза. Если амилоидоз не развивается, то ОПЖ пациентов с TRAPS не отличается от таковой в популяции. В табл. 8 перечислены ДК.
Рисунок 6. Кожные проявления периодического синдрома, связанного с рецептором фактора некроза опухоли: А — правый бок пациента с мутацией Т50М; В — серпигинозная сыпь на лице, шее, туловище и верхних конечностях у ребенка с мутацией C30S; С — эритематозные макулярные высыпания с корками на сгибательной поверхности правой руки у пациента с мутацией Т50М
Почти все TRAPS-ассоциированные мутации локализуются во внеклеточном домене белка TNFR1, при этом 1/3 из них вызвана заменой др. аминокислоты на высококонсервативный остаток цистеина, что приводит к разрыву дисульфидных связей и неправильной укладке белка. Было показано, что ряд др. миссенс-мутаций, не связанных с остатками цистеина, оказывают аналогичное влияние на складывание белка TNFR1. Неправильно сложенный TNFR1 в/клеточно агрегирует, что ведет к конститутивной передаче сигнала через митоген-активируемые протеинкиназы/ядерный фактор (NF)-kB, что, в свою очередь, приводит к высвобождению провоспалительных цитокинов, таких как IL-6, IL-1β и ФНО-α. Замена глутамина на аргинин в остатке 92 (R92Q) и замена лейцина на пролин в остатке 46 (P46L) наблюдается у >1% белого и черного населения соответственно.
Эти варианты не приводят к тем же биохим. и сигнальным нарушениям, которые наблюдаются при более тяжелых TRAPS-мутациях, и, как и в случае с E148Q при FMF, ведутся споры, являются ли они мягкими мутациями/функциональным полиморфизмом.
Колхицин чаще всего неэффективен при TRAPS. При относительно легкой форме заболевания м.б. достаточно НПВП. В более тяжелых случаях с нечастыми приступами м.б. эффективны ГКС в период приступа, но нередко потребность в них со временем растет. Этанерцепт часто эффективно снижает тяжесть и уменьшает частоту обострений, но длительное наблюдение за пациентами с TRAPS, получавшими этанерцепт, показывает, что со временем его эффективность снижается. Следует отметить, что лечение TRAPS моноклональными АТл против ФНО-α иногда приводило к парадоксальному обострению заболевания. У пациентов с TRAPS отмечался «+» ответ на терапию анакинрой, канакинумабом, моноклональным АТл против IL-1β и тоцилизумабом, моноклональным АТл против IL-6. Для пациентов с TRAPS >2 лет одобрено применение канакинумаба, эффективность которого доказана в многоцентровом РКИ*.
P.S. * Канакинумаб в лечении периодической болезни и др. аутовоспалительных заболеваний — исследование CLUSTER, журнал «Клиническая фармакология и терапия», выпуск 2018.4.
4. Криопирин-ассоциированные периодические синдромы. CAPS — спектр заболеваний, в который входят FCAS, MWS и NOMID. Хотя мы и обозначили 3 отдельных клинических диагноза, важно понимать, что в действительности криопиринопатии — единый континуум, разделенный в зависимости от тяжести заболевания. Этот спектр заболеваний обусловлен мутациями в NLRP3 (ранее известном как CIAS1), который кодирует белок под названием криопирин; >100 мутаций NLRP3 при этом заболевании перечислены в онлайн-базе данных Infevers. Достижения в области секвенирования позволили выявлять пациентов с симптомами соматической мозаичности NLRP3.
NLRP3 — белок, содержащий домен PYRIN, который мощно экспрессируется в миелоидных клетках и, в меньшей степени в др. тканях. Он является частью макромолекулярного комплекса, называемого инфламмасомой NLRP3, который активирует про-IL-1β до его зрелой формы в ответ на множество молекулярных паттернов эндогенной угрозой и патоген-ассоциированных молекулярных паттернов. У пациентов с криопиринопатией наблюдаются мутации приобретения функции в NLRP3, которые приводят к конститутивной/легко запускаемой активации инфламмасомы NLRP3. Криопиринопатии проявляются рекуррентными лихорадками и сыпью, напоминающей крапивницу, которая развивается в раннем младенческом возрасте (рис. 7). При гистопатологическом исследовании обнаруживается периваскулярный нейтрофильный инфильтрат без тучных клеток/дегрануляции тучных клеток, наблюдаемых при истинной крапивнице.
Рисунок 7. Уртикарноподобная сыпь. Проявление воспаления и поражения органов при интерлейкин-1-опосредованных заболеваниях: при мультисистемном воспалительном заболевании с началом в неонатальном периоде, представляющем собой тяжелую форму криопи-рин-ассоциированных периодических синдромов и дефицит антагониста рецептора интерлейкина-1. Такая сыпь не является подлинной крапивницей и возникает вследствие инфильтрации кожи нейтрофилами
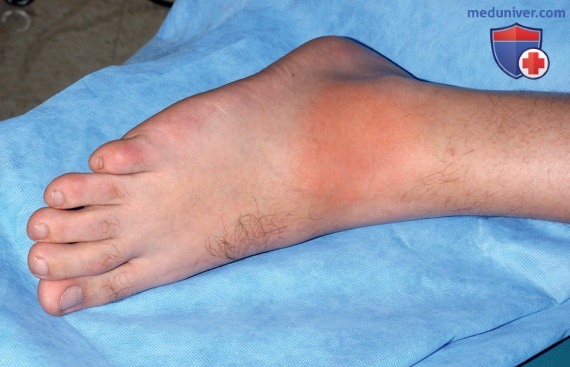
Приступы лихорадки у пациентов с FCAS обычно начинаются через 1-3 ч после пребывания на холоде. Кроме этого, у пациентов с FCAS наблюдается полиартралгия суставов кистей, коленных суставов и лодыжек, также во время приступов может развиваться конъюнктивит. Приступы FCAS купируются самостоятельно и обычно разрешаются в течение 24 ч. АА-амилоидоз при FCAS встречается редко.
В табл. 9 перечислены ДК FCAS.
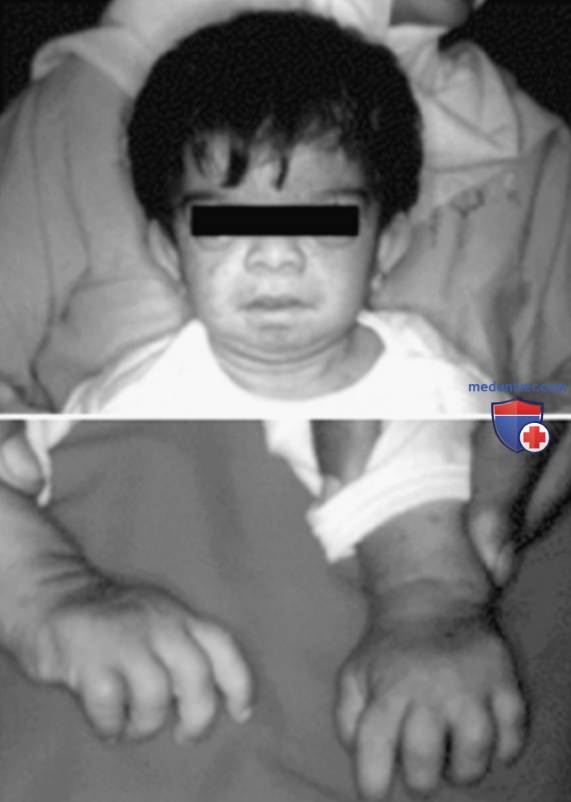
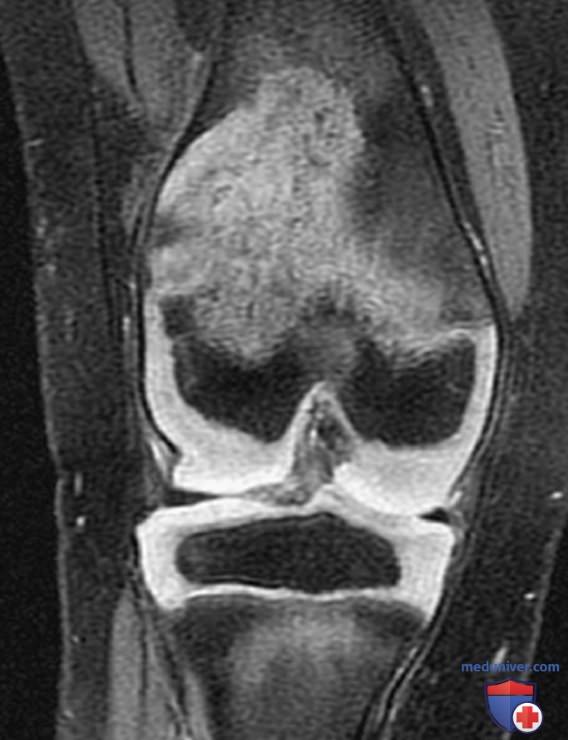
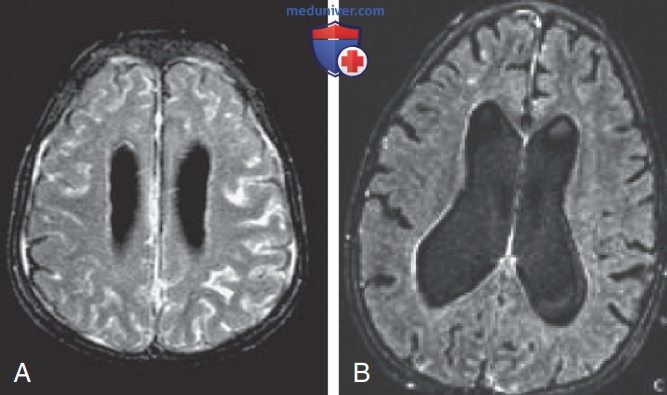
В отличие от FCAS, приступы лихорадки при MWS не провоцируются пребыванием на холоде, но характеризуются такой же сыпью, напоминающей уртикарную, что и при FCAS (рис. 8). У многих пациентов с MWS также развивается прогрессирующая нейросенсорная тугоухость, и при отсутствии лечения у 30% пациентов с MWS развивается АА-амилоидоз. У пациентов с NOMID в неонатальный период отмечаются диффузная уртикарная сыпь, ежедневная лихорадка и дисморфические признаками (рис. 9). Значительные деформации суставов, особенно коленных, могут развиться вследствие чрезмерного разрастания эпифизов длинных костей (рис. 10). У пациентов с NOMID, кроме вышеперечисленного, развивается хронический асептический менингит, который приводит к повышению ВЧД, отеку диска зрительного нерва, ухудшению зрения, прогрессирующей нейросенсорной тугоухости и умственной отсталости (рис. 11).
Рисунок 8. Сыпь на коже, напоминающая крапивницу, у пациента с синдромом Макла-Уэллса
Рисунок 9. 3-летняя девочка с мультисистемным воспалительным заболеванием с началом в неонатальном периоде/хроническим детским неврологическим, кожным и суставным синдромом. Обратите внимание на выраженную деформацию кистей, сыпь, выступающий лоб и большие размеры головы
Рисунок 10. Гипертрофия метафизов. Проявления воспаления и поражения органов при интерлейкин-1-опосредованных заболеваниях: мультисистемном воспалительном заболевании с началом в неонатальном периоде (тяжелая форма криопирин-ассоциированных периодических синдромов) и дефиците антагониста рецептора интерлейкина-1
Рисунок 11. (А) Лептоменингеальное усиление; (В) гидроцефалия и церебральная атрофия. Проявления воспаления и поражения органов при интерлейкин-1-опосредованных заболеваниях: мультисистемном воспалительном заболевании с началом в неонатальном периоде (тяжелая форма криопирин-ассоциированных периодических синдромов) и дефиците антагониста рецептора интерлейкина-1
Таргетная терапия анакинрой (рекомбинантный антагонист IL-1R) изменила жизнь пациентов с NOMID, т.к. этот ЛП позволяет не только контролировать лихорадку и сыпь, но и предотвращать повреждение органов-мишеней. Анакинра, рилрнацепт и канакинумаб эффективны как при FCAS, так и при MWS. Эти ЛП одобрены FDA для лечения обоих заболеваний. Для пациентов с CAPS >2 лет одобрено применение канакинумаба, эффективность которого доказана в многоцентровом РКИ*, анакинра одобрена для детей с 8 мес с МТ >10 кг*. Агрессивная блокада IL-1 привела к уменьшению выраженности амилоидоза при криопиринопатиях.
P.S. * Канакинумаб в лечении периодической болезни и др. аутовоспалительных заболеваний — исследование CLUSTER, журнал «Клиническая фармакология и терапия», выпуск 2018.4.
P.S. ** Инструкция к применению ЛП анакинра.
в) Другие менделевские аутовоспалительные заболевания:
1. Синдром пиогенного артрита с гангренозной пиодермией и акне. PAPA — это редкое АуД-заболевание, вызванное мутациями в PSTPIP1, гене, который кодирует цитоскелетный протеин-1, взаимодействующий с пролин-серин-треонин-фосфатазой (PSTPIP). Белок PSTPIP1 взаимодействует с рядом иммунологически значимых молекул, включая CD2, белком синдрома Вискотта-Олдрича (Wiskott, Aldrich) и пирином. РАРА-ассоциированные мутации PSTPIP1 значительно повышают его сродство к пирину и вызывают повышенную продукцию IL-1β.
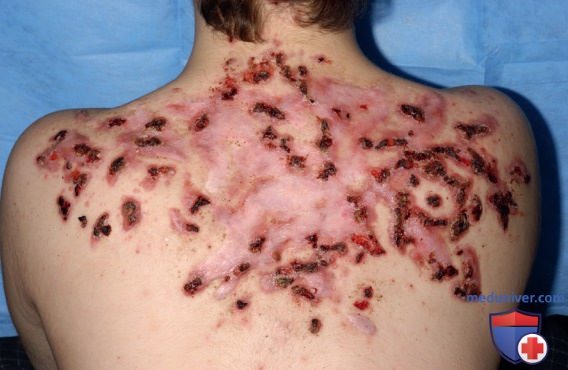
Чаще всего РАРА-синдром впервые клинически проявляется в раннем детстве. Для него характеры повторяющиеся эпизоды стерильного пиогенного артрита, который, по-видимому, развивается спонтанно/после небольшой травмы, и ведет к образованию эрозий и разрушению суставов. Лихорадка в данном случае не является доминирующим признаком. Кожные проявления, как правило, развиваются в подростковом возрасте, такие пациенты склонны к развитию тяжелых кистозных угрей. Кроме того, у пациентов с PAPA обычно развивается гангренозная пиодермия с изъязвлениями (рис. 12), а у некоторых — патергические реакции.
Рисунок 12. Гангренозная пиодермия у пациента с РАРА-синдромом и мутацией А230Т в PSTPIP1. Обратите внимание на диффузные рубцы в верхней части спины, свидетельствующие о предшествующих поражениях
Лечение синдрома PAPA может включать применение ГКС, антагонистов IL-1 и ингибиторов ФНО-α, иногда в комбинации. Суставные проявления PAPA, по-видимому, хорошо отвечают на блокаду IL-1, тогда как кожные, по всей видимости, лучше отвечают на блокаду ФНО-α. Местные процедуры, такие как аспирация суставной жидкости, дренирование полости сустава и интенсивный уход за ранами, так же важны при лечении пациентов с PAPA, как и обезболивание при кожных поражениях. Следует соблюдать осторожность при назначении сульфаниламидов, поскольку у некоторых пациентов с PAPA на фоне их приема развивается панцитопения.
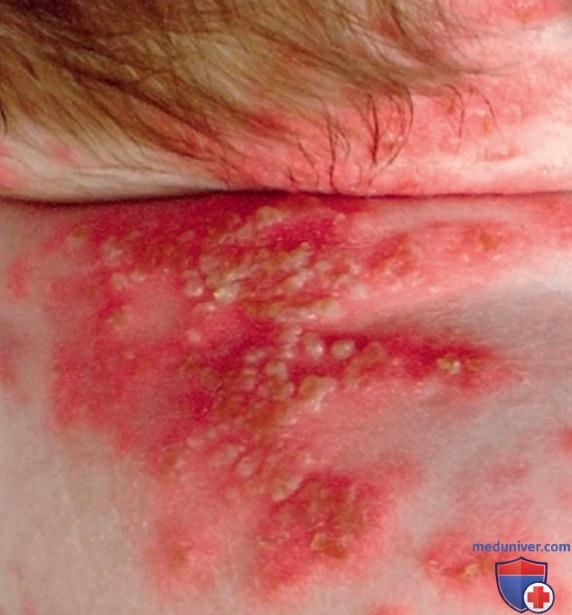
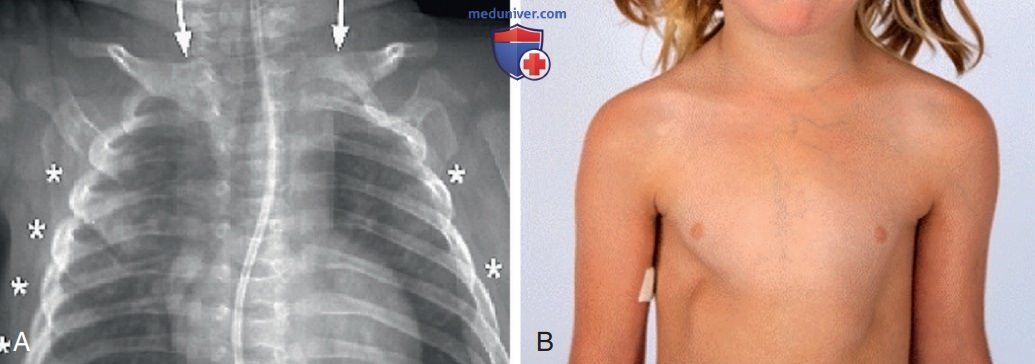
2. Дефицит антагониста рецептора интерлейкина-1. DIRA — АуР аутовоспалительное заболевание, рассматриваемое отдельно от криопиринопатий. DIRA обычно проявляется в неонатальном периоде системным воспалением и нейтрофильным пустулезом, стерильным мультифокальным остеомиелитом, расширением передних концов ребер, периоститом и остеопенией (рис. 13 и 14). И хотя лихорадка не является преобладающим клиническим проявлением DIRA, у пациентов значительно повышены реактанты острой фазы. Возможно развитие СПОН и интерстициального фиброза легких, которые могут привести к летальному исходу.
Рисунок 13. Пустулезная сыпь. Проявления воспаления и поражения органов при интерлейкин-1-опосредованных заболеваниях: мультисистемном воспалительном заболевании с неонатальным началом (тяжелая форма периодических синдромов, ассоциированных с криопирином) и дефиците антагониста рецептора интерлейкина-1. Такую сыпь также можно наблюдать при дефиците антагониста рецептора интерлейкин-36
Рисунок 14. (А) Расширение нескольких ребер (*) и ключиц (стрелки) при остеомиелите при дефиците антагониста рецептора интерлейкина-1; (В) деформация грудной клетки. Проявления воспаления и поражения органов при интерлейкин-1-опосредованных заболеваниях: мультисистемном воспалительном заболевании с неонатальным началом и дефиците антагониста рецептора интерлейкина-1
Развитие DIRA обусловлено мутациями потери функции в IL1RN, кодирующем антагонист IL-1R. Из-за отсутствия антагонистической активности клетки гиперчувствительны к стимуляции IL-1β. Были опробованы многочисленные методы лечения DIRA, включая НПВП, ГКС, в/в Ig, метотрексат, циклоспорин и этанерцепт. Тем не менее анакинра остается ЛП выбора, т.к. он по существу заменяет потерянный белок и обеспечивает развитие быстрого клинического ответа. Анакинра вводится ежедневно, доза титруется для достижения нормального уровня СРБ. В настоящее время существуют анти-IL-1 ЛП длительного действия, канакинумаб и рилонацепт, которые настолько же эффективны и требуют менее частого приема, чем анакинра.
3. Синдром Блау. Синдром Блау — редкое АуД-заболевание, которое проявляется ранним (<5 лет) гранулематозным артритом, увеитом и сыпью. Артрит может поражать лодыжки и запястья и приводить к сгибательным контрактурам пальцев рук и ног (камптодактилии). Саркоидоз с ранним началом проявляется схожей клинической симптоматикой, иногда с поражением внутренних органов. Оба вышеперечисленных состояния обусловлены мутациями в белке 15-го домена рекрутирования каспазы (CARD15), также известном как белок нуклеотид-связывающего домена олигомеризации-2 (NOD2). NOD2 — это в/клеточный сенсор бактериальных продуктов в дендритных клетках, миеломоноцитарных клетках и клетках Панета (Paneth). Мутации в домене олигомеризации NACHT этого белка обуславливают развитие синдрома Блау/раннего саркоидоза, тогда как варианты с мутациями, главным образом, в богатом лейцином повторяющемся домене вызывают повышенную восприимчивость к болезни Крона. ГКС до сих пор остаются основой терапии синдрома Блау, также имеется ряд сообщений о «+» эффектах ингибиторов ФНО-α.
4. Аутовоспаление с дефицитом антител, ассоциированных с фосфолипазой Cγ2 и иммунной дисрегуляцией. APLAID — заболевание, наследуемое по АуД-типу и характеризующееся рецидивирующими поражениями кожи в виде волдырей, бронхиолитом, артралгией, поражением глаз, энтероколитом, отсутствием ауто-АТл и легким иммунодефицитом. Сыпь — первое проявление APLAID, которое описывается как буллезный эпидермолиз по всей поверхности тела. Со временем эта сыпь переходит в рецидивирующие бляшечные и везикулопустулезные поражения, развитие которых провоцируется воздействием тепла и солнечного света. Колит проявляется в возрасте <5 лет. Глазные проявления начинаются в возрасте <1 года и включают язвы и эрозии роговицы, катаракту. К иммунным проявлениям относится заметное снижение уровня IgD-«-» В-клеток памяти, переключенных на синтез др. классов Ig, что ведет к снижению уровней IgM и IgA.
У пациентов с APLAID обнаруживается миссенс-мутация с расширением функции в аутоингибиторной области фосфолипазы Сγ2 (PLCγ2), которая приводит к повышению активности нижележащих медиаторов и стимуляции лимфоцитов. Несмотря на усиление сигнала, конечные популяции иммунных клеток плохо функционируют. Интересно, что др. мутация в комплексе PLCγ2 приводит к развитию синдрома, известного как PLCγ2-ассоциированный дефицит АТл с иммунной дисрегуляцией (APLAID; англ. Autoinflammation with PhosphoLipase Cγ2-associated Antibody deficiency and Immune Dysregulation), который характеризуется холодовой крапивницей и гипогаммаглобулинемией, приводящей к повышенной восприимчивости к инфекциям и аутоиммунным реакциям.
Из-за малого количества описанных пациентов не существует четких схем лечения APLAID. Пациентов лечили НПВП, но м.б. эффективны и ГКС, однако их долгосрочное применение ограничено в виду побочных эффектов. Также с некоторым успехом применялись ингибиторы ФНО-α и ингибиторы IL-1.
5. Дефицит аденозиндезаминазы-2. DADA2 — аутовоспалительное заболевание, вызванное мутациями с потерей функции в гене CECR1, кодирующем аденозиндезаминазу 2-го типа. DADA2 проявляется рецидивирующими лихорадками и спектром сосудистых проявлений, которые включают livedo racemosa*, ранние ишемические лакунарные инсульты и системный васкулит сосудов среднего калибра, похожий на узелковый полиартериит. Лакунарные инсульты, обычно поражающие глубокие ядра и ствол ГМ, возникают в возрасте <5 лет, обычно во время эпизодов воспаления. Во время этих же эпизодов часто наблюдается ливедоподобная сыпь, а при биопсии обнаруживается преобладание нейтрофилов и макрофагов и васкулит сосудов среднего калибра. Обычно повышаются уровни реактантов острой фазы. К др. признакам относят поражение глаз, лимфопению разл. степени, гипогаммаглобулинемию (обычно за счет IgM), гепатоспленомегалию, портальную гипертензию и нейтропению.
Пациенты могут соответствовать ДК узелкового полиартериита, у них может наблюдаться некроз пальцев и феномен Рейно (Raynaud).
P.S. * Livedo racemosa - состояние кожи со стойким красным/фиолетовым обесцвечиванием, характеризующееся ломаным, разветвленным, прерывистым и неправильным рисунком. М.б. ограничено конечностями/быть диффузным.
Аденозиндезаминаза 2-го типа вырабатывается в основном моноцитами и макрофагами, обнаруживается в плазме и, по-видимому, действует как фактор роста и дифференцировки для субпопуляции воспалительных макрофагов. Для лечения пациентов с дефицитом аде-нозиндезаминазы 2-го типа (DADA2; англ. deficiency of adenosine deaminase-2) были опробованы многочисленные противовоспалительные ЛП, включая ГКС и циклофосфамид. В настоящее время основой лечения являются ингибиторы ФНО-α (этанерцепт/адалимумаб), есть отдельные сообщения относительно эффективности анакинры. Основные источники аденозиндезаминазы-2 — макрофаги и моноциты, что повышает возможную роль трансплантации костного мозга в достижении окончательного излечения.
6. Сидеробластная анемия с иммунодефицитом, лихорадкой и задержкой развития. SIFD — это синдром, характеризующийся системным воспалением, лихорадкой, энтеритом и сидеробластной анемией, вызываемый биаллельными мутациями в TRNTI. SIFD проявляется в младенческом возрасте лихорадкой, повышением маркеров воспаления, гастроэнтеритом и анемией. В биоптатах костного мозга обнаруживаются кольцевидные сидеробласты. К др. признакам относят гипогаммаглобулинемию, В-клеточную лимфопению, задержку развития и разл. ВПР НС, судороги и нейросенсорную тугоухость. При визуализации ГМ обнаруживают атрофию мозжечка, задержку миелинизации белого в-ва и снижение перфузии. Прочие изолированные клинические признаки включают: нефрокальциноз, аминоацидурию, ихтиоз, кардиомиопатию и пигментный ретинит. TRNT1 — РНК-полимераза, которая необходима для созревания цитозольных и митохондриальных транспортных РНК, т.к. она добавляет 2 цитозина и 1 аденозин к концам транспортной РНК.
Основа терапии SIFD — симптоматическое лечение с регулярными гемотрансфузиями и заместительной терапией Ig. Перегрузка железом в результате частых гемотрансфузий обычно требует хелатной терапии. У одного пациента анакинра облегчила приступы лихорадки, но никак не повлияла на др. клинические проявления. Среди пациентов с SIFD наблюдается высокая смертность. Одному пациенту в 9 мес была проведена трансплантация костного мозга, благодаря которой удалось скорректировать гематологические и иммунологические нарушения.
7. Дефицит антагониста рецептора интерлейкина-36. DITRA характеризуется эпизодами диффузной эритематозной пустулезной сыпи (генерализованный пустулезный псориаз), лихорадкой, общим недомоганием и системным воспалением. Приступы могут провоцироваться инфекцией, беременностью и менструацией/возникать спонтанно. Генетическая основа этого заболевания — АуР-мутации в гене IL36RN, который кодирует антагонист IL-36R. IL-36 связан с антагонистом IL1R и действует похожим образом, предотвращая выработку воспалительных цитокинов, таких как IL-8. Интересно отметить, что сыпь при DITRA похожа на сыпь при DIRA (дефицит IL-1R, см. ранее), но DITRA зачастую ограничена кожей. DITRA пытались лечить разл. способами, включая аналоги витамина А, циклоспорин, метотрексат и ингибиторы ФНО-α. В отчетах о клинических случаях описано и использование анакинры, которое привело к облегчению симптомов.
8. Семейный холодовой аутовоспалительный синдром 2-го типа. Мутации в NLRP12 приводят к синдрому периодической лихорадки, который характеризуется ТТ >40 °C, артралгиями и миалгиями продолжительностью 2-10 дней. Это расстройство называется FCAS2, т.к. эти эпизоды м.б. спровоцированы пребыванием на холоде. Клинические симптомы могут включать: крапивницу, боль в животе и рвоту, афтозные язвы и лимфаденопатию. Как и при синдроме Макла-Уэллса, описаны случаи развития нейросенсорной тугоухости и неврита зрительного нерва. NALP12 — член семейства белков CATERPILLAR, которые играют важную роль во врожденном иммунитете. Как и toll-подобные рецепторы (TLR; англ. Toll-like receptors), которые распознают молекулярные патогенные паттерны (РАМР; англ, pathogen-associated molecular patterns), NLRP12 способен воспринимать РАМР и приводить к активации воспаления и выработке IL-1β.
Лечение мутаций NALP12 представляло большие трудности до появления антагонистов IL-1 (напр., анакинры), которые на сегодняшний день являются терапией первой линии при FCAS2, и приводят к значительному уменьшению симптомов. Кроме того, некоторую активность демонстрирует колхицин, а системные ГКС способны сократить длительность приступа.
9. Аутовоспаление с энтероколитом. Это заболевание, вызванное мутациями в NLRC4, характеризующееся развивающимся во младенчестве энтероколитом, лихорадкой и эпизодами аутовоспаления. Маркеры воспаления, включая СРБ и ферритин, чаще всего бывают повышены. Синдром активации макрофагов, характеризующийся панцитопенией, гипертриглицеридемией и коагулопатиями, часто встречается во время обострений, которые могут провоцироваться эмоциональным и физическим стрессом. Также часто отмечаются рецидивирующие миалгии с эпизодами лихорадки. Это заболевание вызвано миссенс-мутациями с расширением функции в NOD-подобном рецепторе С4 (NLRC4; англ. NOD-like receptors С4), который в норме способствует активации инфламмасомы. Образующийся в результате белок приводит к конститутивной продукции IL-1. Основой терапии являются антагонисты IL-1, такие как анакинра, канаки-нумаб и рилонацепт. До постановки диагноза пациенты с мутациями NLRC4 с переменным успехом лечились колхицином и ГКС внутрь.
10. Синдром Маджида. Синдром Маджида — это АуР-заболевание, вызванное мутациями в гене LPIN2 (см. табл. 4). Клинически синдром Маджида впервые проявляется в детском возрасте рецидивирующими лихорадками, стерильным остеомиелитом, врожденной дизеритропоэтической анемией, нейтрофильным дерматозом, нарушениями нормального развития и гепатомегалией. Лечение синдрома Маджида включает НПВП, ГКС и антагонист IL-1R. Как именно мутации в LPIN2 приводят к аутовоспалительному заболеванию, до сих пор точно не известно.
11. Интерферонопатии. IFN типа 1 (IFN-α и IFN-β) — первая линия защиты от вирусных инфекций, они вырабатываются разл. типами клеток. Во время вирусных инфекций вирус производит множество продуктов, включая ss-PHK, ds-PHK, и CpG-содержащую ДНК, которые распознаются в/клеточными сенсорами. Затем эти сенсоры индуцируют продукцию IFN типа 1, которая активирует рецепторы IFN и IFN-чувствительные гены, чтобы помочь контролировать распространение вируса до тех пор, пока не будет активирована адаптивная иммунная система для элиминации вируса. Неправильная активация этих путей приводит к продукции IFN и интерферонопатиям.
12. Хронический атипичный нейтрофильный дерматоз с липодистрофией и повышенной температурой. CANDLE, также известный как протеасома-ассоциированный аутовоспалительный синдром/синдром суставных контрактур, мышечной атрофии и липодистрофии, индуцированной панникулитом — АуР-заболевание. Пациенты обращаются в раннем возрасте по поводу рецидивирующей лихорадки и явлений системного воспаления, поражений кожи, включая кольцевидную эритему, узловатую эритему, панникулит и нейтрофильный дерматоз, небольших контрактур суставов, липодистрофии, атрофии мышц/миозита, фиолетовой отечности век и анемии. Часто встречаются конъюнктивит, асептический менингит и органомегалия. Повышены уровни реактантов острой фазы и количество тромбоцитов. Могут возникать аутоиммунные реакции, включая Кумбс (Coombs)-«+» гемолитическую анемию и гипотиреоз. Интеллект и развитие обычно не нарушаются, хотя были сообщения о незначительных задержках развития.
CANDLE вызывается мутациями потери функции в PSMB8, гене, кодирующем субъединицу β5i протеасомы. Протеасомы играют важную роль в деградации убиквитинированных белков, что обеспечивает надлежащий гомеостаз белков, их дефекты приводят к клеточному стрессу и высвобождению воспалительных цитокинов, включая IFN 1-го типа.
На сегодняшний день не существует разработанных схем лечения CANDLE-синдрома, хотя были предприняты попытки использовать разные методы терапии, включая колхицин, дапсон, циклоспорин, инфликсимаб и этанерцепт, все с минимальным успехом. При лечении ГКС и метотрексатом удавалось немного уменьшить выраженность симптомов. Эффективность анакинры не подтвердилась, в то же время блокаторы IL-6 продемонстрировали некоторую эффективность. Поскольку рецепторы IFN используют путь JAK/STAT (англ. Janus kinases, signal transducer and activator of transcription proteins — янус-киназы, преобразователь сигнала и активатор транскрипционных белков) для передачи сигнала, есть надежда на ЛП группы ингибиторов JAK (тофацитиниб, руксолитиниб и барицитиниб).
13. STING-ассоциированная васкулопатия с началом в младенческом возрасте. SAVI — редкое заболевание, которое проявляется в младенческом возрасте. Оно обусловлено мутациями в гене ТМЕМ173, который кодирует стимулятор генов IFN (STING; англ. stimulator of interferon genes — стимулятор генов интерферона). К ранним проявлениям относят системное воспаление с лихорадкой и повышенными маркерами воспаления. Поражение кожи включает нейтрофильную сыпь и фиолетовые высыпания на коже пальцев рук, ног, носа, щек и ушей. Эти поражения со временем прогрессируют и могут приобрести некротический характер вследствие окклюзии сосудов. При гист. исследовании поражений обнаруживается дермальное воспаление с лейкоцитокластическим васкулитом и микротромботической ангиопатией. Поскольку STING также экспрессируется в легочном эпителии, у пациентов с SAVI развивались осложнения со стороны легких, включавшие паратрахеальную аденопатию, интерстициальное заболевание легких и фиброз.
STING является адаптерным белком механизма распознавания в/клеточной ДНК, и опосредует выработку IFN-β. Затем IFN-β передает сигнал через рецептор IFN, активируя сигнальный путь JAK/STAT и нижележащие IFN-чувствительные гены, включая IL-6 и ФНО-α. Мутации STING, вызывающие SAVI — мутации de novo с расширением функции, которые активируют спонтанную продукцию IFN-β.
Вариантов лечения пациентов с SAVI в настоящее время немного, хотя недавние данные, касающиеся ингибиторов JAK (тофацитиниб, руксолитиниб и барицитиниб), продемонстрировали многообещающие результаты в части блокирования передачи сигналов рецептора IFN-β и активации генов IFN-ответа.
1. Периодическая лихорадка, афтозный стоматит, фарингит и аденит. PFAPA — наиболее частый синдром рекуррентной лихорадки у детей. Обычно он проявляется в 2-5 лет повторяющимися эпизодами лихорадки, недомогания, экссудативного тонзиллита с «-» посевами из зева, шейной лимфаденопатией, афтами в полости рта и, реже, головной болью, болью в животе и артралгией. Эти эпизоды длятся 4-6 дней, независимо от приема жаропонижающих ЛП/АБ, и часто возникают «по часам», с 3-6-недельной цикличностью. Во время этих эпизодов может обнаруживаться небольшая гепатоспленомегалия, лейкоцитоз и повышенное содержание реактантов острой фазы. С возрастом частота и интенсивность приступов уменьшаются. Этиология и патогенез PFAPA остаются неизвестными.
У большинства пациентов наблюдается резкий ответ на однократный прием преднизолона внутрь (0,6-2,0 мг/кг), но нужно заметить, что этот подход не предотвращает развитие рецидивов и может сократить интервал между обострениями. Примерно в трети случаев для предотвращения развития рецидивов эффективен циметидин 20-40 мг/кг в сутки. Небольшие серии исследований показали, что во время обострений м.б. эффективной ана-кинра, но, учитывая эффективность ГКС, ее применение не рентабельно. Колхицин может увеличить время между приступами. Были сообщения о полном излечении после тонзиллэктомии, однако основным подходом остается медикаментозное лечение.
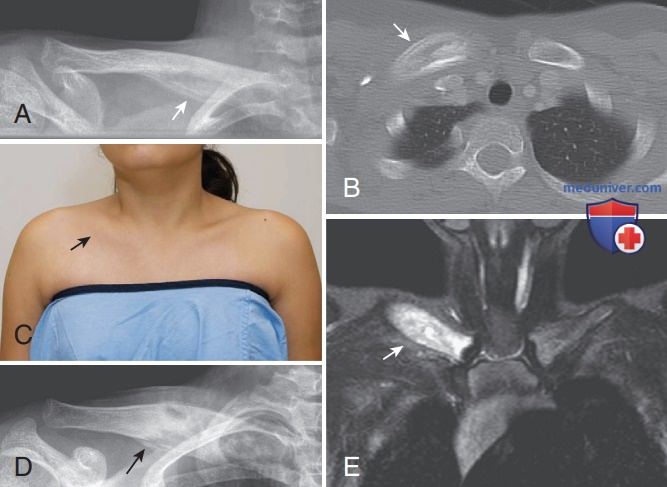
2. Хронический рецидивирующий мультифокальный остеомиелит. Хронический рецидивирующий мультифокальный остеомиелит — это форма воспалительного заболевания костей, чаще всего встречающаяся у детей (см. табл. 4). Гистологически и радиологически этот остеомиелит практически неотличим от инфекционного остеомиелита (рис. 15). Пациенты обычно жалуются на боли в костях, также могут отмечаться лихорадка, отек мягких тканей и повышение уровней реактантов острой фазы. Культуральные посевы стерильны. Наиболее часто вовлекаемые кости: дистальный отдел бедренной кости, проксимальный отдел большеберцовой/малоберцовой кости, кости позвоночника и таза. Наблюдаются как метафизарные, так и эпифизарные поражения, также может развиваться преждевременное закрытие зон роста. Реже поражаются ключицы и нижняя челюсть. ДД необходимо проводить с инфекционным остеомиелитом, гистиоцитозом и ЗНО (нейробластомой, лимфомой, лейкозом, саркомой Юинга (Ewing)).
Рисунок 15. Поражение ключицы при хроническом рецидивирующем мультифокальном остеомиелите. Девушка-подросток с односторонним поражением ключицы. (А) На рентгенограмме правой ключицы обнаруживается расширение медиальных двух третей с соответствующей периостальной реакцией. (В) При последующей компьютерной томографии правой ключицы визуализируется расширение медиальной части правой ключицы с участками склероза, окруженными зонами периостальной реакции (стрелка). (C) Обострение болезни через 18 мес, при осмотре заметно дальнейшее увеличение ключицы (фото из клинического архива). (D) На рентгенограмме правой ключицы через 18 мес наблюдаются выраженный интервальный склероз и утолщение. (E) В это же время на МРТ обнаруживается повышенная интенсивность сигнала на Т1-взвешенных изображениях правой медиальной ключицы с подавлением жировой ткани и контрастированием, что отражает продолжающееся воспаление
Синовит, акне, пустулез, гиперостоз и остит (SAPHO*), возможно, являются эквивалентом хронического рецидивирующего мультифокального остеомиелита у взрослых. Этиология спорадических случаев неизвестна. Этот остеомиелит наблюдается при синдроме Маджида (см. ранее) в сочетании с ВЗК и воспалительным заболеванием кожи, таким как ладонно-подошвенный пустулез. ЛП стартовой терапии являются НПВП. Терапия второй линии включает применение ГКС, ингибиторов ФНО и бисфосфонатов.