Диагностика, патология, наследственность и течение мукополисахаридозов
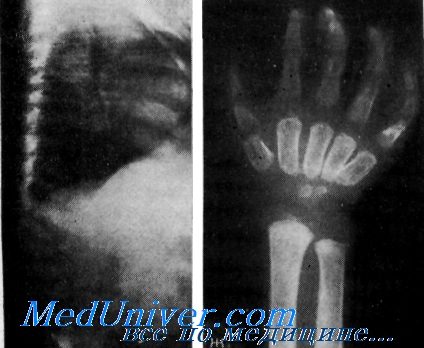
Рентгенологически при всех формах МПС, кроме синдрома Моркио, выявляются выраженные в различной степени множественные дизостозы. Наиболее тяжелые формы наблюдаются при синдромах Гурлер (МПС-1-Г) и Марото — Лами (МПС-VI). Череп больных становится большим и деформированным, турецкое седло принимает У-образпую форму. Ребра становятся широкими, а тела позвонков — диспластичными с двояковыпуклыми концевыми пластинками. Тела позвонков нижнегрудного и верхнепоясничного отделов после 12—18 мес жизни приобретают крючковидную конфигурацию. В длинных трубчатых костях отмечаются расширение диафизов, искривление и деформация эпифизов.
Диафизы коротких трубчатых костей пережаты. Фаланги и II—V пястные кости имеют пулевидную форму.
Рентгенологические изменения при синдроме Шейе незначительны. Отмечаются когтеобразная деформация пальцев и маленькие обильные кисты в костях запястья.
При синдроме Хантера (МПС-II) отмечаются менее резко выраженные, чем при синдроме Гурлер, множественные дизостозы.
При синдроме Санфилиппо (МПС-III) свод черепа утолщается позже, сосцевидный отросток склерозирован, а тела позвонков имеют яйцевидную форму. Руки нормальные.
Рентгенологические изменения при синдроме Моркио (МПС-IV) включают: платиспопдилшо, гипоплазию зубовидного отростка, вальгусную деформацию бедер, расширение подвздошной кости и дисплазию головки бедра. Основания II—V пястных костей имеют исчерченный вид, а кости запястья малы и имеют неправильную форму.
Установлены дефектные энзимы. При синдромах Гурлер и Шсйе отмечается недостаточность a-L-идуронидазы. Оба типа синдрома Хантер характеризуются недостаточностью сульфоидуронат-сульфатазы. При синдроме Санфилиппо (МПС-III-А) имеется недостаточная активность гепаран-N-сульфатазы, при МПС-III-Б — недостаточность N-ацетил-b-D-глюкозаминидазы. При МПС-VI отмечается дефект арилсульфатазы-В. Различные синдромы характеризуются избыточным выделением с мочой следующих мукополисахаридов: МПС-1 — дерматан-сульфата и гепараи-сульфата, главным образом первого, МПС-II — дерматан-сульфата и гспаран-сульфата в равных количествах, МПС-III — гепараи-сульфата, МПС-IV — кератап-сульфа-та, МПС-VI — дерматан-сульфата.
Наследственность. За исключением синдрома Хантер, при котором наследование осуществляется Х-сцепленным путем, все другие рассмотренные на нашем сайте мукополисахаридозы наследуются по аутосомно-рецессивному типу.
Диагноз. Мукополисахаридозы чаще всего путают с муколипидозами. В последней группе заболеваний не наблюдается ни накопления кислых мукополисахаридов, ни выделения их с мочой. Дифференциальный диагноз среди классических мукополисахаридозов очень труден. У больных с синдромами Гурлер и Хантер обычно наблюдается умственная отсталость, но на первом году жизни они выглядят вполне полноценными. При синдроме Марото — Лами умственное развитие больных нормальное, но отмечается помутнение роговицы. При синдроме Хантер роговицы совершенно чистые.
Лечение. Предложить эффективную длительную терапию при мукополисахаридозах не представляется возможным.
Прогноз. Смотри выше, в обсуждении.
Выводы. Общая характеристика этих синдромов включает: 1) аутосомно-рецессивное наследование, кроме Х-сцепленного синдрома Хантер; 2) грубые черты лица и множественные дизостозы, кроме синдрома Моркио; 3) накопление и выделение с мочой одного и более специфических мукополисахаридов и 4) глухоту, различающуюся по типу и степени тяжести.