Почечные изменения при синдроме Альпорта демонстрируют комбинированные черты хронического гломерулонефрита, пиелонефрита и интерстициального нефрита, но с недостаточно полной характеристикой каждого из этих заболеваний.
Почки маленьких размеров имеют мелкозернистое и палисадное строение. На срезе поверхности коры могут быть обнаружены желтые линейные прожилки. Первичные изменения встречаются в соединительнотканной мембране клубочков.
Наиболее ранним поражением являются выраженная пролиферация эпителия в клубочках, интерстициальный фиброз или локальное расширение с атрофией канальцев, появляющиеся приблизительно одновременно. В большинстве случаев наблюдаются заполненные липидами пенистые клетки, не связанные с эпителиальными клетками канальцев. Эти пенистые клетки могут заполнять интерстициальную ткань, особенно в нижних отделах коры, располагаясь в виде рядов или гнезд. В большинстве случаев в интерстициальной ткани находят плазматические клетки, лимфоциты и отложения кальция. Капальцевыс изменения включают атрофию эпителия и расширение некоторых канальцев. Иногда наблюдается хронический интерстициальный нефрит, при котором изначально мало клубочков (White et al., Krickstein et al.).
Необходимо подчеркнуть, что, в то время как все эти изменения обусловлены синдромом Альпорта, они не являются специфичными для него. Исследование ультраструктуры установили характерное расслоение соединительнотканной мембраны клубочков па большое число тонких пластинок в результате накопления между слоями маленьких плотных частичек (Churg a. Sherman, Scherman et al.).
Имеется несколько сообщений, касающихся патологии височной кости при синдроме Альпорта. Winter с сотр. обнаружили интактность всех структур, кроме утраты примерно 50% клеток спирального узла в базальном завитке улитки. Gregg и Becker, а также Ваbai и Beltez описали дегенеративные изменения сосудистой полоски и волосковых клеток кортиева органа, главным образом в базальном завитке улитки. Fujita и Hayden, Westergaard с соавт., Myers и Tyler, Bergstrom с сотр. не нашли характерных для синдрома Альпорта изменений височной кости.
Патогенез. Патогенез синдрома Альпорта неизвестен. Quick с соавт., Arnold и Weidaver отметили сходство улитки и почек по некоторым пунктам (баланс жидкости и электролитов, общие ототоксические и нефротоксические вещества). Они продемонстрировали иммунохимические и иммуногистохимическис доказательства сходства антигенов в почках и в улитке.
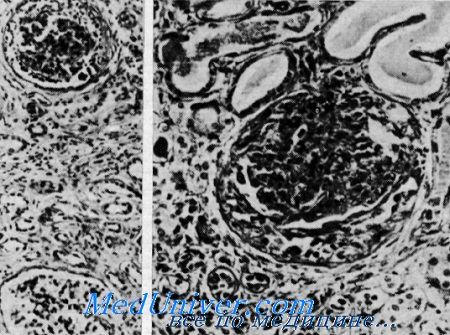
Нефрит и нейросенсорная глухота (синдром Альпорта). Сверху клубочек с утолщенной основной мембраной и слипшейся капсулой в противоположность почти нормальному клубочку внизу. Видны редкие клубочки, имеющие полулупиую форму, расширенные канальцы, содержащие белковые цилиндры. Утолщенная основная мембрана, окружающая атрофичные канальцы в области фиброза (из H. I. Krickstein et al.).
Наследственность. В некоторых исследованиях было высказано предположение, что гетерозиготные матери передают аномальный ген примерно 65% своих детей. Среди потомства больных мужчин синдром Альпорта отмечается у 75% их дочерей и только у 45% их сыновей. Cohen с соавт., Shaw и Glover, MacNeill и Shaw предположили, что синдром определяется одним аутосомным геном, который регулярно наследуется у женщин (имея сродство скорее к овоцитам, чем к полярным тельцам) и наследуется преимущественно с Х-хромосомой при сперматогенезе.
Preus и Fraser предположили, что неблагоприятная внутриутробная среда больной матери может быть причиной повышенной ненетрантности гена у ее сыновей. Мы согласны с Мауо в том, что синдром Альпорта, вероятно, генетически гетерогенен.
Диагноз. Синдром Альпорта должен быть отграничен от всех других случаев гематурии и иротеинурии в детском возрасте. Гематурией сопровождаются также гломерулопефрит и гломерулит. Наследственный нефрит без глухоты, описанный Dockhorn, является, по-видимому, другим заболеванием.
Лечение. Глухота обычно уменьшается при пользовании слуховыми аппаратами. Терапия должна быть направлена на коррекцию биохимических нарушений, сопровождающих почечную недостаточность.
Необходимо обсудить вопрос о пересадке почек. У одного больного в результате этой операции мы наблюдали улучшение слуха.
Прогноз. Прогноз варьирует. У некоторых больных заболевание может протекать легко и практически не оказывать влияния на их жизнь, в то время как у других больных может развиться тяжелая почечная недостаточность, от которой они погибают в молодом возрасте.
Выводы. Характеристика этого синдрома включает: 1) аутосомнодоминантное наследование с более тяжелым поражением мужчин; 2) прогрессирующий нефрит с уремией; 3) патологию хрусталика, включающую лентиконус, сферофакию или катаракты, примерно в 10% случаев; 4) начинающуюся в нервом или во втором десятилетии жизни прогрессирующую пейросенсорную глухоту с варьирующей экспрессивностью.