Синдромы Лоренса-Муна и Барде-Бидля - клиника, диагностика
Синдромы Лоренса-Муна и Барде-Бидля - заболевания с аутосомно-рецессивным наследованием. Клиническая картина складывается из ожирения, пигментной дегенерации сетчатки, гипогонадизма, умственной отсталости, а также полидактилии (при синдроме Барде—Билля) или спастической параплегии (при синдроме Лоренса—Муна).
Иногда выявляются кисты почек. Морфологическая картина напоминает таковую при нефронофтизе.
Для этих синдромов характерна локусная генетическая гетерогенность: заболевание может развиться в результате мутации одного из четырех генов — BBS1, BBS2, ARL6(BBS3) или BBS4, расположенных в сегментах 11q13, 16q21, 3р12—3ql3 и 15q22.3— 15q23 соответственно. Прямой анализ ДНК при этом заболевании пока недоступен.
Подробнее синдром разбирается в статье из раздела клинических синдромов.
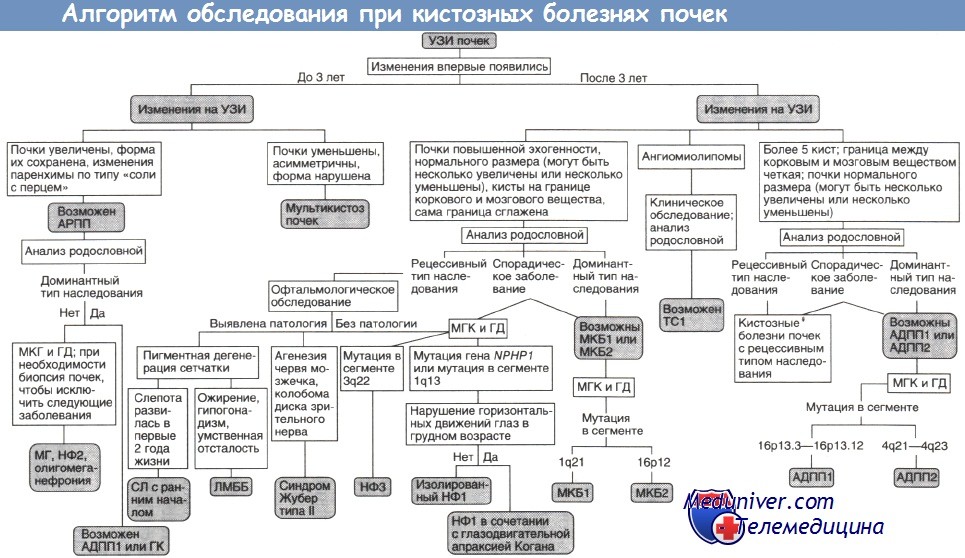
АДПП1, АДПП2 — аутосомно-доминантныи поликистоз почек типов I и II;
АРПП — аутосомно-рецессивный поликистоз почек;
ГД — генодиагностика (при необходимости ГД включают в МГК);
ГК — гломерулокистоз почек;
ЛМББ — синдромы Лоренса—Муна и Барде—Бидля;
МГ — синдром Меккеля—Грубера;
МГК — медико-генетическое консультирование;
МКБ1, МКБ2 — медуллярная кистозная болезнь типов I и II;
НФ1, НФ2, НФЗ — нефронофтиз типов I, II и III;
СЛ — синдром Сениора—Локен;
TC1 — туберозный склероз типа I.