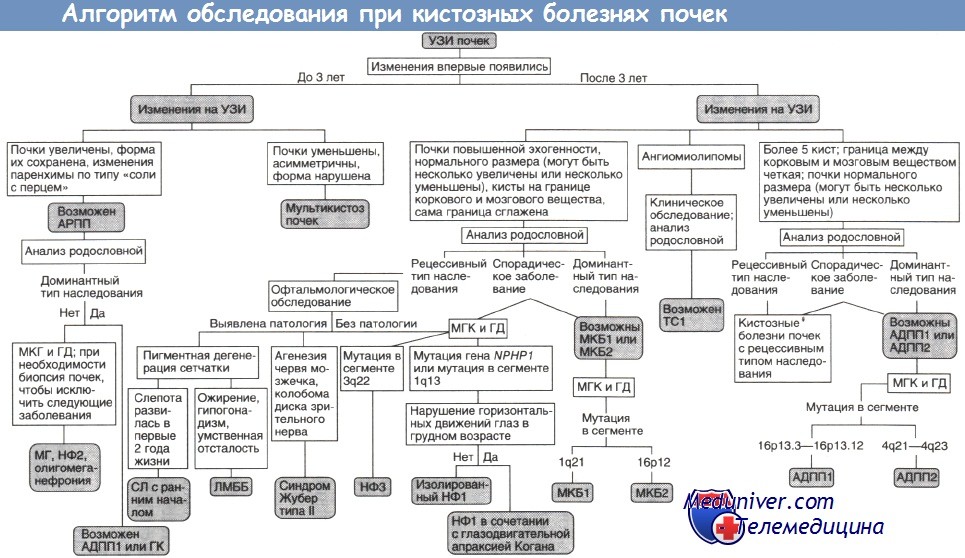
Выделяют три формы нефронофтиза — все с аутосомно-рецессивным типом наследования. Нефронофтиз типа II начинается в грудном возрасте, нефронофтиз типа III впервые проявляется у подростков, а начало нефронофтиза типа I (ювенильного нефронофтиза) приходится на препубертатный период. Иногда ювенильный нефронофтиз протекает с внепочечными проявлениями.
Нефронофтиз типа I (ювенильный нефронофтиз)
Для детей с ювенильным нефронофтизом характерны полиурия и полидипсия, анемия и задержка роста. Поскольку заболевание не проявляется типичными признаками гломерулопатии (гематурией, протеинурией, артериальной гипертонией, отеками), почечную недостаточность нередко обнаруживают случайно при обследовании по поводу анемии, полиурии или сонливости. Диагноз нефронофтиза в таких случаях может оказаться полной неожиданностью. Средний возраст развития терминальной почечной недостаточности — 13 лет.
Внепочечные проявления нефронофтиза. Ювенильный нефронофтиз может сопровождаться врожденной глазодвигательной апраксией Когана, при которой у гомозиготных по делециям гена NPHP1 детей младше 2 лет отсутствуют произвольные движения глаз по горизонтали. Вместе с пигментной дегенерацией сетчатки нефронофтиз входит в состав синдрома Сениора—Локен, а вместе с агенезией червя мозжечка и колобомой диска зрительного нерва - в состав синдрома Жубер типа II. Описан также нефронофтиз в сочетании с фиброзом печени и с конусовидными эпифизами фаланг.
Наконец, ювенильный нефронофтиз может быть одним из проявлений синдромов Жена (асфиктическая дисплазия грудной клетки) и Эллисаван Кревельда (хондроэктодермальная дисплазия), синдрома RHYNS (Retinitis pigmentosa, HYpopituitarism, Nephronophthisis, mild Skeletal dysplasia — пигментная дегенерация сетчатки, гипопитуитаризм, нефронофтиз, легкие аномалии развития скелета), а также синдромов Лоренса—Муна и Барде—Бидля.
Диагностика нефронофтиза. Важный метод диагностики нефронофтиза — УЗИ, при котором выявляются сглаженность границы между корковым и мозговым веществом, повышенная эхогенность почечной паренхимы, а у детей старше 9 лет — кисты, расположенные на границе коркового и мозгового вещества. Сцинтиграфия обнаруживает снижение концентрирующей способности почек.
Нефронофтиз типа II
Этот тип нефронофтиза называют инфантильным, так как первые его проявления возникают еще во внутриутробном периоде, сразу после рождения или в первый год жизни ребенка. Недавно при исследовании родословной большой семьи бедуинов было установлено, что ген нефронофтиза типа II (NPHP2) находится на 9-й хромосоме (сегмент q22-q31).
Поскольку морфологическая картина и течение этого заболевания довольно сильно отличаются от двух других форм нефронофтиза, возможно, что в группу нефронофтиза оно попало не совсем обоснованно.
Нефронофтиз типа III
Ген нефронофтиза типа III (NPHP3) идентифицирован при исследовании еще одной большой родословной — на этот раз семьи из Венесуэлы. Этот тип нефронофтиза называют подростковым. Терминальная почечная недостаточность наступает на 6 лет позже, чем у больных ювенильным нефронофтизом, — в среднем в 19 лет. В остальном же ни морфологической картиной, ни клиническими проявлениями нефронофтиз типа III от ювенильного нефронофтиза не отличается.
Генодиагностика нефронофтиза
Идентификация гена NPHP1, ответственного за ювенильный нефронофтиз, позволила использовать для диагностики прямой анализ ДНК. Продукт этого гена нефрокистин содержит SH3-домен, что, по-видимому, указывает на участие нефрокистина в межбелковых взаимодействиях, в том числе во внутриклеточной передаче сигнала в фокальных контактах — участках, в которых клетка соприкасается с внеклеточным матриксом.
Нарушением фокальных контактов могут объясняться характерные разрывы базальной мембраны почечных канальцев, наблюдаемые при нефронофтизе. У 85% детей делеции обнаруживаются на обоих аллелях гена NPHP1. На выявлении этих делеций, а также точечных мутаций гена NPHP1 основывается генодиагностика нефронофтиза. Гомозиготные делеции гена NPHP1 описаны также при врожденной глазодвигательной апраксии Когана и при поздней форме пигментной дегенерации сетчатки.
При глазодвигательной апраксии Когана детям грудного и младшего возраста не удаются горизонтальные движения глаз и, чтобы посмотреть в сторону, они совершают резкие толчкообразные движения головой.
АДПП1, АДПП2 — аутосомно-доминантныи поликистоз почек типов I и II;
АРПП — аутосомно-рецессивный поликистоз почек;
ГД — генодиагностика (при необходимости ГД включают в МГК);
ГК — гломерулокистоз почек;
ЛМББ — синдромы Лоренса—Муна и Барде—Бидля;
МГ — синдром Меккеля—Грубера;
МГК — медико-генетическое консультирование;
МКБ1, МКБ2 — медуллярная кистозная болезнь типов I и II;
НФ1, НФ2, НФЗ — нефронофтиз типов I, II и III;
СЛ — синдром Сениора—Локен;
TC1 — туберозный склероз типа I.