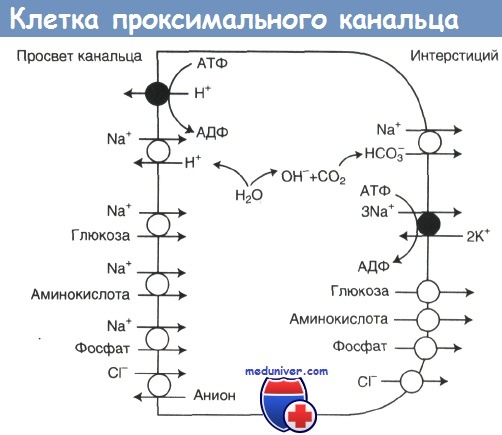
Отфильтровавшиеся в клубочках аминокислоты реабсорбируются в проксимальных канальцах. Как и в случае глюкозы, перенос аминокислот через апикальную мембрану осуществляется путем котранспорта с натрием, а перенос через базолатеральную мембрану — путем облегченной диффузии.
Аминоацидурия может возникать:
1) при повышении СКФ и избыточном поступлении аминокислот в проксимальные канальцы;
2) при врожденных нарушениях метаболизма, приводящих к повышению концентрации определенных аминокислот в крови;
3) при нарушении реабсорбции аминокислот вследствие дефекта белков-переносчиков.
Цистинурия — самое частое нарушение канальцевого транспорта аминокислот. Она наследуется аутосомно-рецессивно и вызвана мутацией гена SLC3A1, кодирующего белок — переносчик диаминомонокарбоновых аминокислот (лизина, аргинина, орнитина и цистина). Экскреция всех этих аминокислот увеличена. Из-за плохой растворимости цистина в моче он образует мочевые камни.
Камни возникают в любом возрасте, но чаще между 10 и 30 годами. Рецидивы мочекаменной болезни вызывают нарастающее повреждение почек.
Цистинурию следует заподозрить у любого больного с рецидивами мочекаменной болезни. Экскреция цистина с мочой более 250 мг на 1 г креатинина подтверждает диагноз цистинурии. В моче видны характерные шестиугольные кристаллы цистина. Лечение направлено на предупреждение образования мочевых камней.
Важно, чтобы больной пил много жидкости, не допуская повышения концентрации цистина в моче. Ощелачивание мочи (рН выше 6,5—7) повышает растворимость цистина, но избыточное ощелачивание повышает риск образования фосфатных камней. Некоторым больным показаны комплексобразующие средства: тиопронин или пеницилламин.
Иминоглицинурия — аутосомно-рецессивное заболевание, при котором нарушен канальцевый транспорт глицина, пролина и гидроксипролина. У некоторых больных нарушено также всасывание этих аминокислот в кишечнике. В отличие от гиперпролинемии, гидроксипролинемии, а также от глицинемии без кетоза при иминоглицинурии уровень этих аминокислот в плазме остается в норме. Заболевание доброкачественное, лечение не требуется.
Хартнуповская болезнь — нарушение транспорта моноаминомонокарбоновых аминокислот в почках и тонкой кишке. Уровень этих аминокислот в плазме в норме или понижен. Заболевание наследуется аутосомно-рецессивно, у большинства больных оно протекает бессимптомно, но у некоторых возникают симптомы пеллагры: дерматит по типу солнечного ожога на открытых участках кожи, атаксия и поведенческие расстройства.
К этому приводит дефицит никотиновой кислоты из-за нарушения ее всасывания в кишечнике и повышенного выведения триптофана с мочой. Больным назначают никотинамид.
Клетка проксимального канальца; показаны переносчики апикальной и базолатеральной мембран. Транспорт большей части растворенных веществ через апикальную мембрану является Na+-зависимым. Движущей силой для этого транспорта служит трансмембранный концентрационный градиент Na+, создаваемый за счет выкачивания Na+ из клетки Na+,К+-АТФазой базолатеральной мембраны. Выход растворенных веществ через базолатеральную мембрану в основном осуществляется за счет облегченной диффузии, не зависящей от Na+.