Этиология и встречаемость болезни Тея-Сакса. Болезнь Тея-Сакса (MIM №272800), раннедетский ганглиозидоз GM2, — панэтническое аутосомно-рецессивное заболевание распада ганглиозидов, вызванное недостаточностью гексозаминидазы А (см. главу 12). Кроме раннедетской тяжелой формы, недостаточность гексозаминидазы А вызывает легкую форму болезни с началом в юношеском или взрослом возрасте.
Встречаемость недостаточности гексозаминидазы А широко варьирует в различных популяциях; встречаемость болезни Тея-Сакса в Северной Америке колеблется от 1 на 3600 новорожденных у евреев ашкенази до 1 на 360 000 среди не ашкенази евреев. Сравнимую с евреями-ашкенази встречаемость болезни Тея-Сакса имеют французские канадцы, каджуны в Луизиане и амиши в Пенсильвании. Повышенная частота носительства в этих четырех популяциях — следствие генетического дрейфа, хотя не исключено преимущество гетерозигот.
Патогенез болезни Тея-Сакса
Ганглиозиды — церамидовые олигосахариды, присутствующие в поверхностных мембранах всех клеток, но больше всего их в клетках мозга. Ганглиозиды концентрируются в поверхностных мембранах нейронов, особенно в аксонах и дендритах. Они функционируют как рецепторы различных гликопротеиновых гормонов и бактериальных токсинов и задействованы в дифференцировке клеток и межклеточном взаимодействии.
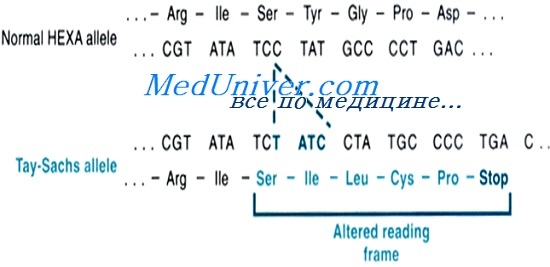
Гексозаминидаза А — лизосомный фермент, состоящий из двух субъединиц. а-Субъединица кодируется геном НЕХА в хромосоме 15, а бета-субъединица — геном НЕХВ в хромосоме 5. В присутствии белка-активатора гексозаминидаза А удаляет концевой N-ацетилгалактозамин из ганглиозида GM2.
Мутации генов а-субъединицы или белка-активизатора вызывают накопление GM2 в лизосомах и, этим самым, раннедетский, позднедетский или взрослый тип болезни Тея-Сакса. [Мутация а-субъединицы вызывает болезнь Сандхоффа (MIM № 268800)].
Механизм того, как накопление ганглиозида GM2 вызывает смерть нейронов, полностью не определен, хотя, по аналогии с болезнью Гоше, нейропатологию могут вызывать токсичные побочные продукты ганглиозида GM2. Уровень остаточной активности гексозаминидазы А обратно пропорционален тяжести болезни.
Пациенты с раннедетской формой ганглиозидоза GM2 имеют два патологических аллеля, приводящих к полному отсутствию активности гексозаминидазы. Пациенты с формами ганглиозидоза GM2 с началом в юношеском или взрослом возрасте — обычно сложные гетерозиготы по аллелю с полным отсутствием функции и аллелю с небольшой остаточной активностью гексозаминидазы А.
Фенотип и развитие болезни Тея-Сакса
Раннедетский тип ганглиозидоза GM2 характеризуется неврологическим ухудшением, начинающимся в возрасте 3-6 мес и приводящим к смерти к 2-4 года. Обычно моторное развитие останавливается или начинает регрессировать к 8-10 мес жизни, и в течение второго года жизни развивается неспособность к самостоятельному передвижению. Потеря зрения начинается на первом году жизни и быстро прогрессирует; почти всегда это связано с «вишнево-красным» пятном («косточкой») при обследовании глазного дна.
Судороги обычно начинаются в конце первого года жизни и непрерывно становятся все тяжелее. Дальнейшее ухудшение на втором году жизни заканчивается децеребрационной ригидностью, затруднениями глотания, тяжелыми судорогами и, наконец, вегетативным состоянием.
Ганглиозидоз GM2 с позднедетским началом выявляют на 2-4 годах жизни, он характеризуется неврологической симптоматикой, начинающейся с атаксии и дискоординации. К концу первого десятилетия большинство пациентов имеют спастичность и судороги; к 10-15 годам у большинства развивается децеребрационная ригидность и вегетативное состояние со смертью обычно во втором десятилетии жизни. Снижение зрения отмечают, но может не быть «вишневой косточки» на глазном дне; атрофия зрительного нерва и пигментный ретинит часто появляются в конце течения болезни.
Взрослый тип ганглиозидоза GM2 имеет выраженную клиническую изменчивость (прогрессирующая дистония, спиноцеребеллярная дегенерация, патология моторных нейронов или психиатрические нарушения). До 40% больных имеют прогрессирующие психиатрические проявления без психоза. Зрение затрагивается редко, и данные офтальмологического обследования обычно в норме.
Особенности фенотипических проявлений болезни Тея-Сакса:
• Возраст начала: от раннего детства до взрослого возраста
• Нейродегенерация
• «Вишневая косточка»
• Психоз
Лечение болезни Тея-Сакса
Диагноз ганглиозидоза GM2 ставят на основании выявления как отсутствующей или почти отсутствующей активности гексозаминидазы А в сыворотке крови или в лейкоцитах, так и нормальной или повышенной активности гексозаминидазы В. Для диагностики также можно использовать анализ мутаций в гене НЕХА, но обычно его выполняют только для уточнения транспортного носительства и пренатальной диагностики.
Болезнь Тея-Сакса в настоящее время — инкурабельное заболевание; следовательно, лечение направлено на устранение симптомов и паллиативный уход. Почти все больные требуют фармакологического лечения судорог. Психиатрические проявления пациентов с взрослым типом ганглиозидоза GM2 обычно не поддаются стандартным антипсихотическим или противодепрессивным средствам; наиболее эффективны препараты лития и электросудорожная терапия.
Риски наследования болезни Тея-Сакса
Для потенциальных родителей без GM2-ганглиозидоза в семейном анамнезе эмпирический риск родить ребенка, больного СМ2-ганглиозидозом, зависит от частоты заболевания в их этнических группах. Для большинства жителей северной Америки эмпирический риск носительства составляет приблизительно 1 на 250-300, но для евреев-ашкенази эмпирический риск носительства — приблизительно 1 на 30. Для пары, в которой оба родителя носители, риск родить ребенка с ганглиозидозом GM2 равен 1/4.
Пренатальная диагностика основана на идентификации мутаций в гене НЕХА или на определении недостаточности гексозаминидазы А в тканях плода, например, ворсинах хориона или амниоцитах. Для эффективной идентификации пораженного плода с помощью анализа мутации в гене НЕХА обычно необходимо, чтобы мутации, вызывающие ганглиозидоз GM2 в семье, уже были известны.
Скрининг популяций высокого риска на носительство и последующие превентивные мероприятия уменьшили встречаемость болезни Тея-Сакса среди евреев-ашкенази почти на 90%. По традиции такой скрининг выполняют по определению активности гексозаминидазы А сыворотки крови с искусственным субстратом.
Этот чувствительный метод, тем не менее, не способен различить патологические мутации и псевдонедостаточность (снижение распада искусственного субстрата, но нормальный распад естественного субстрата); следовательно, носительство обычно подтверждают молекулярным анализом НЕХА. В гене НЕХА обнаружено два аллеля псевдонедостаточности и более 70 патологических мутаций.
Среди евреев-ашкенази, положительных по результатам ферментного скрининга, 2% — гетерозиготны по аллелю псевдонедостаточности, и 95-98% гетерозиготны по одной из трех патологических мутаций, две вызывают раннедетскую форму, одна — взрослую форму GM2-ганглиозидоза. В отличие от этого, среди остальных североамериканцев, положительных по результатам ферментативного скрининга, 35% — гетерозиготны по аллелям псевдонедостаточности.
Пример болезни Тея-Сакса. Семейная пара, оба евреи ашекенази, направлена в клинику генетики для оценки риска родить ребенка с болезнью Тея-Сакса. Сестра жены умерла от болезни Тея-Сакса в детстве. Дядя мужа по отцу находится в психиатрической лечебнице, но его диагноз неизвестен. Как муж, так и жена отказались от скрининга на носительство болезни Тея-Сакса в подростковом возрасте.
Анализ фермента показал, что как муж, так и жена имеют чрезвычайно низкую активность гексозаминидазы А. Последующий молекулярный анализ мутаций, преобладающих у евреев ашкенази, подтвердил, что у жены имеется патогенная мутация, тогда как у мужа только аллель псевдонедостаточности.