Болезнь Вильсона у детей. Болезнь Галлервордена-Шпатца
Болезнь Вильсона - редкое (1:40 000-1:100 000 живых новорожденных) наследственное заболевание с аутосомно-рецессивным типом наследования, представляет собой врожденное нарушение транспорта меди и характеризуется развитием цирроза печени и дегенеративными изменениями в ЦНС, особенно в базальных ганглиях. Ген болезни Вильсона (WND) картирован на хромосоме 13 в локусе 13ql4-21. Различные мутации гена определяют вариабельность клинических проявлений заболевания.
Точная причина болезни Вильсона неизвестна. Основной механизм его развития связывают с уменьшением экскреции меди с желчью, что частично обусловлено дефектом лизосом гепатоцитов. Первые проявления заболевания у детей до 10 лет связаны с острой или подострой печеночной недостаточностью, которая часто ошибочно диагностируется как инфекционный гепатит. Неврологические проявления болезни Вильсона редко возникают до 10 лет, первым симптомом часто служит прогрессирующая дистония.
Затем появляется тремор в конечностях. Вначале он односторонний, но в конечном итоге становится грубым, генерализованным и приводит к нарушению двигательной активности пациента (так называемый порхающий тремор). Симптомы прогрессирующего поражения базальных ганглиев включают слюнотечение, насильственную улыбку, вызванную ретракцией верхней губы, дизартрию, Дисфонию, ригидность, контрактуры, дистонию и хореоатетоз. Кольцо Кайзера-Флейшера, которое лучше всего определяется при обследовании Щелевой лампой, считается патогмоничным симптомом в результате отложения меди в десцемето-Вой оболочке.
В отсутствие лечения развивается деменция, пациенты утрачивают способность к самостоятельному передвижению (прикованы к постели), через несколько лет после дебюта заболевания развивается кома с летальным исходом. На МРТ и КТ в развернутой стадии заболевания Расширение желудочков, атрофия вещества мозга, поражение таламуса и базальных ганглиев.
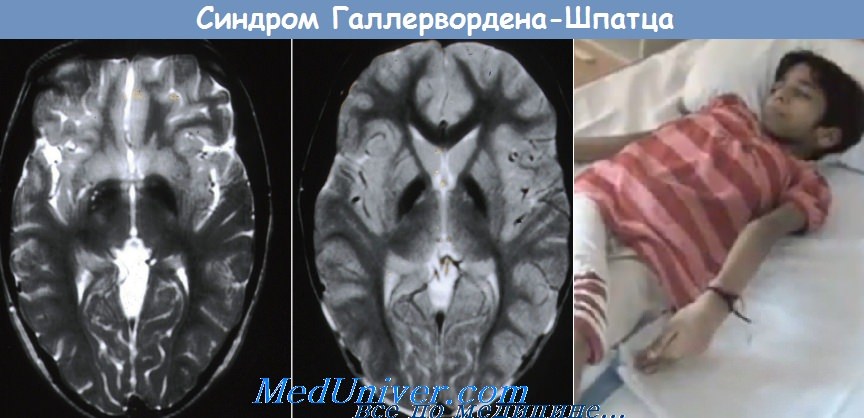
Болезнь Галлервордена-Шпатца
Болезнь Галлервордена-Шпатца — редкое дегенеративное заболевание с аутосомно-рецессивным типом наследования. Ген картирован на хромосоме 20 в локусе 20р13. Заболевание обычно начинается в детском возрасте и характеризуется прогрессирующей мышечной дистонией, ригидностью и хореоатетозом. Спастичность, положительный симптом Бабинского, дизартрия и признаки деменции появляются в подростковом возрасте; заболевание обычно заканчивается летальным исходом в молодом возрасте. На МРТ поражение бледного шара, включая зону пониженной интенсивности сигнала в Т2-взвешенных изображениях (соответствует зоне накопления железосодержащих пигментов) и область повышенной интенсивности сигнала в переднемедиальной области (соответствует зоне вакуолизации) — характерный симптом «глаз тигра».
Патоморфологическое исследование определяет избыточное накопление железосодержащих пигментов в области бледного шара и черного вещества.
В последнее десятилетие достигнуты значительные успехи в лечении дистонии. У детей с генерализованной дистонией, в том числе у пациентов с нарушением глотания, терапевтический эффект может быть получен при применении больших доз тригексифенидила (артан). Начальная доза составляет 2 мг/сут с дальнейшим медленным повышением до 60-80 мг/сут или до появления неблагоприятных побочных эффектов (задержка мочи, спутанность сознания или нечеткость зрения). К дополнительным препаратам, эффективным в лечении дистонии, относятся карбамазепин, леводопа, бромокриптин и диазепам.
При сегментарной дистонии, например при спастической кривошее (тортиколис), часто эффективны инъекции ботулотоксина. Интратекальное введение баклофена через имплантируемый инфузионный насос может быть эффективно у некоторых пациентов. Стереотаксические операции и электростимуляция таламуса и бледного шара с помощью введения глубоких электродов также вызывают хороший эффект у детей с тяжелой дистонией.