Спиноцеребеллярные дегенерации с аутосомно-доминантным наследованием (ADCAS): спиноцеребеллярные атаксии (SCA)
Спиноцеребеллярные дегенерации с аутосомно-доминантным наследованием (ADCAS) включают очень большую группу неврологических заболеваний, проявляющихся атаксией и другими неврологическими симптомами. Известно множество различных типов, отчасти определены дефектные гены. Заболевания чаще всего поражают взрослых, но могут начинаться рано в детском или подростковом возрасте.
Это обстоятельство связано с тем, что данные нарушения в большей или меньшей степени характеризуются феноменом генетической антиципации в сочетании с возрастающим количеством триплетных повторов внутри пораженного гена. Клинические проявления включают прогрессирующую атаксию и различные варианты сочетанных аномалий, таких как офтальмопарез, атрофия зрительного нерва, ригидно-акинетический синдром, поражение черепных нервов и утрата ощущения позы и вибрационной чувствительности. Могут отмечаться легкие расстройства интеллекта. Возможна периферическая нейропатия (Bennett et al„ 1984).
Среди патологоанатомических изменений отмечается прогрессирующая мозжечковая атрофия и в большинстве случаев поражение спинного мозга. Во многих случаях в дегенеративный процесс вовлекаются нижние оливы и ядро ствола мозга. Такие случаи обычно классифицируются в разделе оливопонтоцеребеллярных атрофий (ОРСА). Тем не менее, выделение группы ОРСА имеет только патологоанатомическое обоснование, не согласующееся с клиническими и генетическими классификациями, используемыми в данной статье на сайте.
Более того, к данной группе относится ряд заболеваний с различными причинами, распространенность повреждений при которых может варьировать в широком диапазоне в рамках одной нозологической единицы, а в некоторых случаях отмечается изолированное или явно преобладающее поражение коры мозжечка. Данная группа включает спиноцеребеллярные атаксии и ряд редких синдромов с различным генетическим наследованием, дегенеративные изменения при которых отмечаются часто, но не всегда.
Спиноцеребеллярные атаксии (SCA). Аутосомно-доминантные мозжечковые атаксии характеризуются прогрессирующими мозжечковыми расстройствами координации. Они составляют большую группу заболеваний, к которой в настоящее время относят 28 изученных типов нарушений и множество еще неизученных состояний. Некоторые из этих заболеваний сочетаются с повтором тринуклеотидов в генах, кодирующих полиглутамин.
Некоторые тринуклеотиды транслируются в длинные цепи полиглутамина (polyQ), который в виде включений накапливается в ядрах и/или цитоплазме нейронов. Цепи полиглутамина могут влиять на внутриклеточный транспорт за счет создания помех в метаболических путях (Gunawardena и Goldstein, 2005) или приводят к нарастанию токсического действия (van de Warrenburg et al., 2005). При других типах повторы не транслируются и не сочетаются с наличием полиглутаминовых включений. Механизм действия таких повреждений различен и может включать ингибирование трансляции гена (Everett и Wood, 2004; Duenas et al., 2006) и другие неясные воздействия.
Спиноцеребеллярные атаксии (SCA) чаще всего встречаются у взрослых, и только некоторые из них отмечаются у детей, что обычно является следствием феномена антиципации. Несмотря на варьирование фенотипов различных SCA (Schols et al., 1997), отмечается большое количество совпадений, и чаще всего диагноз только на основании клинических признаков невозможен. Лечение в настоящее время только симптоматическое.
В данном разделе кратко описаны только те формы, которые хотя бы изредка встречаются у детей (SCA1, SCA3 и SCA7).
Понтоцеребеллярная атрофия у двухлетней девочки с задержкой умственного развития, атипичным пигментным ретинитом и атаксией.
Ствол мозга аномально тонкий и отсутствует выступ моста. Червь мозжечка относительно маленький.
1. Спиноцеребеллярная атаксия 1 типа (SCA1). Данное заболевание является следствием экспансии повтора нестабильного тринуклеотида ЦАГ на хромосоме 6р22-р23 (Giunti et al, 1994). SCA1 отмечалось у детей в возрасте 12 лет и старше. Наряду с SCA3 данное заболевание является наиболее частой формой ADCA1 в Западной Европе (Durr et al, 1996b). Однако распределение отдельных форм очень варьирует внутри различных этнических групп. Данное заболевание часто встречается у взрослых, у детей регистрируются только редкие случаи. Симптомы обычно появляются в возрасте 20-40 лет, но встречается феномен антиципации, что приводит к проявлению заболевания у детей.
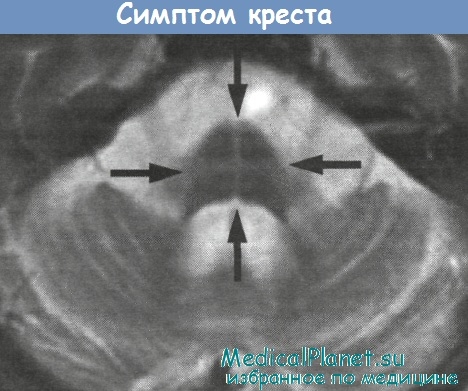
ADCA1 включает несколько генетически изолированных нозологических единиц (SCA 1-6 типов с очень похожими клиническими проявлениями (Rosenberg, 1995). В большинстве случаев на МРТ выявляется заметная атрофия мозжечка, затрагивающая преимущественно червь, и атрофия ствола мозга, в частности моста, нормальное расширение которого исчезает (Wullner et al., 1993).
Симптом креста при спиноцеребеллярной атаксии 1 типа (SCA1)
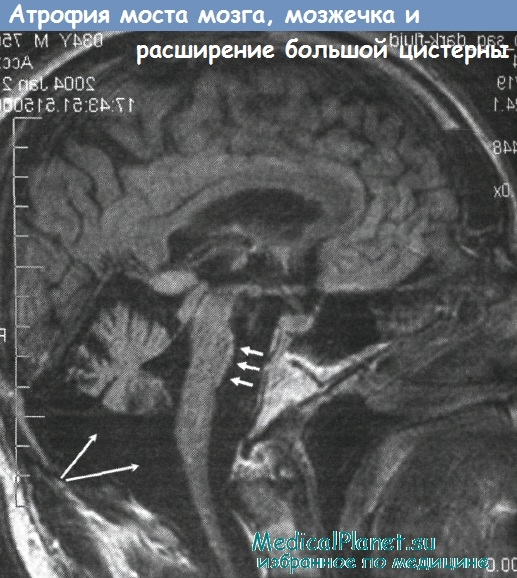
2. Спиноцербеллярная атаксия 7 типа (CSA7). Данное состояние ранее описывалось как OPSA3 и ADCA2. У взрослых отмечается картина прогрессирующей мозжечковой атаксии с дегенерацией макулы. Последнее проявление может длительное время оставаться изолированным. Заметная антиципация может отмечаться при семейных случаях, возможно очень раннее начало (в возрасте от 6 месяцев) (Enevoldson et al., 1994). Такие случаи характеризуются стремительным течением и наступлением смерти. Заболевание может развиться у детей, рожденных клинически здоровыми родителями; в связи в высокой антиципацией при данном заболевании, примерно в 10% случаев у младенцев или детей симптомы появляются раньше, чем у родителей (Enevoldson et al., 1994; Gouw et al., 1994). Ген картирован на хромосоме 3р12-р21.1 (Benomar et al., 1995) и кодирует белок атаксин 7. Включения в клеточных ядрах содержат как атаксин, так и убиквитин (Ansorge et al., 2004).
3. Другие спиноцеребеллярные атаксии с доминантным типом наследования. В таблице ниже указаны гены и их локализация на хромосомах при наиболее частых SCA, также отмечена связь с трансляцией polyQ цепей или ее отсутствие. Мутации локуса SCA3 являются причиной болезни Мачадо-Джозефа (Durr et al., 1996b), которая наряду с SCA1 является частой формой заболевания в Западной Европе. Также отмечается экспансия повторов тринуклеотида ЦАГ (Tuite et al., 1995). Болезнь Мачадо-Джозефа редко встречается у детей. Клинические проявления варьируют.
У детей заболевание часто начинается со спастичности и акинезии или даже дистонии, а в некоторых случаях проявляется преимущественно офтальмоплегией, иногда с мозжечковой атаксией в сочетании с периферической нейропатией или без нее (Giunti et al., 1995). Данное заболевание встречается не только у выходцев с Азорских островов, как считалось изначально. Отмечаются редкие случаи изолированной мозжечковой атаксии.
Атрофия моста, мозжечка при спиноцеребеляррной атаксии 7 типа (SCA7)
4. Сцепленная с Х-хромосомой спиноцеребеллярная атаксия. Зарегистрировано небольшое количество случаев передачи заболевания сцеплено с Х-хромосомой, большая часть — у взрослых пациентов. В небольшом числе семей сцепленная с Х-хромосомой мозжечковая дегенерация начиналась в младенческом или подростковом возрасте. Разница между данными редкими формами спиноцеребеллярной атаксии и паренхиматозной церебеллярной атаксией практически отсутствует (в случаях, когда не проводилось посмертное патологоанатомическое исследование). В действительности в рамках одной родословной могут встречаться случаи без поражения ствола мозга и спинного мозга.
5. Другие редкие синдромы, возможно связанные с SCA и другими редкими дегенеративными заболеваниями. Нозологическое определение некоторых редких заболеваний до сих пор неясно. Данная группа заболеваний включает случаи с выявленным семейным вариабельным сочетанием SCA с дисфункцией задних столбов, голубыми гистиоцитами в спинном мозге, глухотой, гипотонией, низким ростом, умеренной отсрочкой развития, атрофией и миоклонусом.
Прочие синдромы проявляются сочетанием мозжечковой атаксии с экстраневрологической патологией. К данной группе относятся случаи сочетания с болезнью почек (Badhwar et al., 2004), синдромом идиопатического несахарного диабета и атаксии (Birnbaum et al., 1989), случаи атаксии и гипер- и гопогонадотропного гипогонадизма и других разнообразных проявлений (DeMichele et al., 1990; Toscano et al., 1995), включая остеосклероз. Некоторые случаи атаксии с поражением внутренних органов у детей могут относиться к группе синдрома дефицита углеводов гликопротеина (CDG).
Случаи рано начинающейся семейной мозжечковой дегенерации, зарегистрированные Harding и Brett (1995), затрагивают преимущественно клетки Пуркинье и пути зрительного нерва. Группа семейных врожденных гипоплазий до сих пор нечетко классифицирована (al Shahwan et al., 1995). Такие случаи, вероятно, сочетаются с известным генетическим риском семейных рецидивов среди пациентов с атаксическим церебральным параличом.