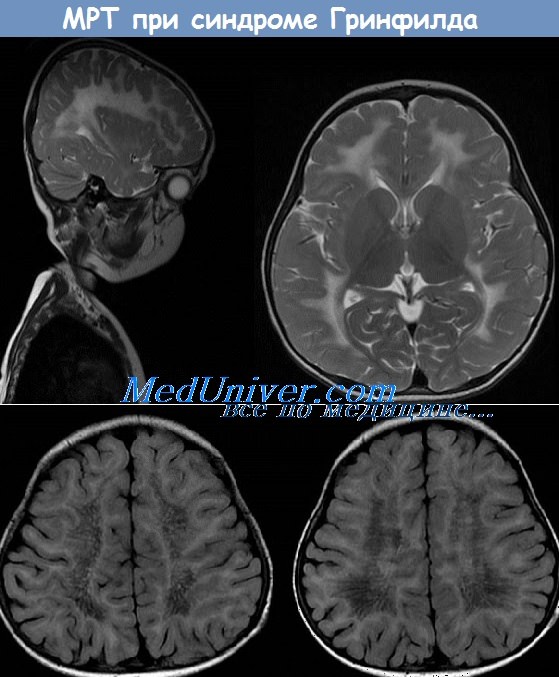
Метахроматическая лейкодистрофия наследуется по аутосомно-рецессивному типу и характеризуется недостаточностью арилсульфатазы А. вследствие чего происходит накопление сульфатидов в головном мозге, периферической нервной ткани и других тканях, например в почках. Чаще всего наблюдается поздняя инфантильная форма. Начиная со 2-ю года жизни развиваются спастические парезы, ухудшаются когнитивные функции, снижаются сухожильные рефлексы, развиваются бульбарные и псевдобульбарные симптомы, в частности дизартрия, а также атрофия зрительного нерва и слепота.
В терминальной сталии заболевания формируется тетраплегия с последующим исходом в вегетативное состояние. Ювенильная норма начинается между 3-м и 10-м годами жизни, в большинстве случаев с нарушений ходьбы, но иногда с эмоциональных расстройств и дсмениии. При взрослой форме, в среднем развивающейся в возрасте около 30 лет. первоначально отмечаются психические нарушения или деменция. спастичность, атаксия. На Т2-взвешспных МРТ-изображсниях выяатяются сливные гиперинтенсивные очаги в белом веществе мозжечка и больших полушарий, атрофия коры, расширение желудочков. Подкорковые структуры остаются в начальных фазах заболевания сохранными.
Уровень арилфосфатазы А снижен в лейкоцитах и моче. Затормозить или остановить прогрессирование заболевания можно с помощью трансплантации костного мозга.
Краббе связана с недостаточностью галактоцереброзидазы. Основные клинические проявления — спастические парезы, атрофия зрительного нерва и снижение скорости проведения по периферическим нервам. Инфантильная форма заканчивается смертью в течение первых 2 лет жизни. Описаны также ювенильная и взрослая формы, симптомы которых подобны проявлениям метахроматической лейкодистрофии.
Мукополисахаридозы представляют собой группу заболеваний, проявляющихся гаргоилизмом, они характеризуются накоплением кислых мукополисахаридов, обмен которых нарушен вследствие недостаточности гидролизы, необходимой для их угилизации.
Синдром Гурлер — наиболее тяжелая форма мукополисахарилоза. Пациенты заболевают в младенческом возрасте и в большинстве случаев умирают до достижения 1 0 лет. На 1 -м году жизни формируются поясничный горб и помутнение роговицы. Суставы становятся ригидными и отечными, грудная клетка деформируется, руки и ноги остаются маленькими и неуклюжими, а с возраста 2—3 лет отмечаются карликовый рост и умственная отсталость. Черты лица грубые, обращают на себя внимание выступающий вперед лоб, густые брови, седловидный нос, гипертелоризм, «бугристый» язык.
Moгут также выявляться утолщение мозговых оболочек, гилроцсфалия или компрессия спинного мозга с тетрапарезом. Причиной смерти нередко становится поражение сердца. Форму заболевания с относительно более благоприятным течением представляет синдром Шейе.
К ним относятся синдромы Гунтера, Санфилиппо, Моркио и Марата-Лами. Синдром Гунтера наследуется по Х-спепленному типу. При синдроме Санфилиппо наблюдается прежде всею поражение головною мозга, а при синдромах Моркио и Марото-Лами преобладают скелетные аномалии.
МРТ при метахроматической лейкодистрофии детей - синдроме Гринфилда