• CFTR представляет собой анионный канал, транспортирующий Cl- или НСO3-
• Нарушение секреторной функции при кистозном фиброзе затрагивает многие органы
Ионные каналы, переносчики и насосы выполняют в клетке много различных функций, нарушение которых служит причиной развития ряда заболеваний. Болезни, развивающиеся при нарушении функционирования ионных каналов, называются каналопатии. Мутации в генах, кодирующих белки ионных каналов, вызывают различные дефекты, которые служат причиной патофизиологических нарушений, наблюдающихся при том или ином заболевании.
В предыдущих статьях мы рассматривали различные каналопатии, которые сопровождались нарушениями транспортной функции почек или секреторной функции различных органов, приводящими к заболеваниям костной, нервной или мышечной ткани, а также к развитию аритмий и к внезапной смерти.
В данной статье мы рассмотрим кистозный фиброз (муковисцидоз), который является типичным заболеванием ионных каналов и патогенез которого становится более понятным по мере выяснения причин нарушений транспортных процесов в мембране эпителиальных клеток.
Кистозный фиброз представляет собой одно из наиболее распространенных генетических заболеваний, сопровождающихся смертельным исходом, которое связано с рецессивными мутациями в гене, кодирующем белок анионного канала. Этот канал называется регулятором трансмембранного переноса при кистозном фиброзе (CFTR). Канал функционирует как АТФ-зависимый анионный канал. Белок экспрессируется в апикальной мембране эпителиальных клеток и может фосфорилироваться при участии протеинкиназы A.
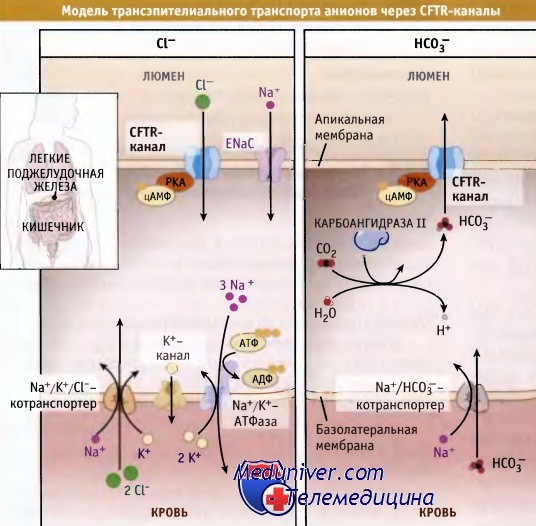
Трансэпителиальный транспорт хлорид и бикарбонат ионов через CFTR-каналы.
CFTR функционируют поблизости от эпителиальных Na+-каналов (ENaCs) апикальной мембраны и являются их ингибиторами.
Снижение функции GFTR приводит к увеличению реабсорбции Na+ и воды через эпителиальные клетки.
CFTR является представителем большой группы переносчиков, которые содержат АТФ-связывающую кассету (АВС). АВС-переносчики представляют собой АТФ-зависимые насосы, способные транспортировать через мембрану различные субстраты, например аминокислоты, пептиды, ионы, сахара, токсины, липиды, а также лекарственные препараты. Наряду с кистозным фиброзом, мутации в гене ABC-переносчиков вызывают развитие ряда других заболеваний, например болезни иммунной системы, развитие устойчивости к антибиотикам и противораковым препаратам.
CFTR переносят через мембрану Cl- или НСO3-. Транспорт каналами ионов Cl- продемонстрирован в экспериментах с выделенными и реконструированными канальными белками, заключенными в ли-посомы. Функционирование CFTR регулируется циклическим АМФ (цАМФ), который представляет собой внутриклеточный вторичный мессенджер. При повышении уровня цАМФ происходит активация протеинкиназы А и фосфорилирование CFTR. При этом активируется транспорт анионов через CFTR. Трансэпителиальный транспорт ионов СЕ через CFTR-каналы происходит за счет Na+-градиента, который поддерживается Na/K+-АТФазой и эпителиальными Na+-каналами (ENaCs), расположенными в апикальной мембране.
CFTR-каналы создают входящий ток ионов Cl-, который вместе с ENaCs обеспечивает электронейтральный транспорт ионов Na+ и Cl- через мембрану. Находящийся в базолатеральной мембране Na+/K+/Cl--котранспортер обеспечивает транспорт в клетку ионов Cl- после гормональной стимуляции.
CFTR экспрессируются в апикальной мембране различных эпителиальных клеток, включая клетки кишечника, легких, поджелудочной и потовых желез. Для функционирования этих органов необходима секреция жидкости. В легком, например, тонкий слой жидкости, увлажняющей эпителиальные клетки, выстилающие воздухоносные пути, необходим для нормального функционирования ресничек. Последние очищают слизь, которая захватывает из воздуха посторонние частицы и бактерии. Эта жидкость образуется при участии CFTR, и они также контролируют ее состав.
У больных кистозным фиброзом слизь отличается повышенной вязкостью, что препятствует ее очистке от патогенов, и поэтому бронхи более подвержены инфекции. В результате накопление вязкой слизи приводит к расстройству дыхания. Блокада вязкой слизью желудочно-кишечного тракта, поджелудочной железы и печени служит причиной расстройства пищеварения и плохого усвоения питательных веществ. Функционирование CFTR при секреции жидкости из апикальной мембраны предполагает, что этот регулятор работает вместе с другими транспортными белками апикальной и базолатеральной мембран эпителиальных клеток. Функционирование CFTR играет важную роль в секреции жидкости, поскольку транспорт солей (в виде ионов Na+ и Cl-) создает осмотическую движущую силу для переноса воды через мембраны.
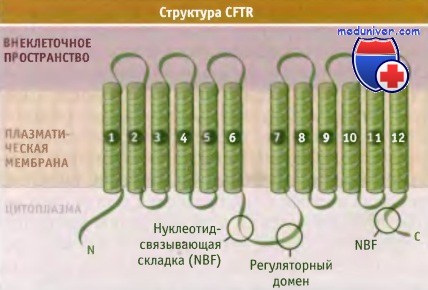
Предполагаемая структура анионного канала,
регулятора трансмембранного переноса при кистозном фиброзе (CFTR).
CFTR-белок содержит две нуклеотид-связывающих складки (NBFs) и уникальный регуляторный домен (R), который находится в цитоплазме, а также два трансмембранных домена. Каждый из трансмембранных доменов состоит из шести трансмембранных сегментов, и два домена вместе с доменом, связывающим нуклеотид, образуют тандемную повторяющуюся структуру. Каждый из нуклеотид-связывающих доменов содержит сайт взаимодействия с АТФ, состоящий из фосфат-связывающей петли, образованной мотивом Уолкера А, одинаковым для многих АТФ-связывающих белков, включая F1F0-АТФ-синтазы.
С возникновением кистозного фиброза связаны сотни различных мутаций в CFTR гене, но наиболее распространенной является делеция, приводящая к дефектному белку, в котором отсутствует Phe508, обычно расположенный в N-терминальном участке NBF. Из-за дефекта перемещения белок CFTRAPhe508 не транспортируется к плазматической мембране и остается в эндоплазматическом ретикулуме.
Поскольку CFTR является каналом, ионы транспортируются в направлении электрохимического градиента без энергетических затрат. Зачем CFTR связывает АТФ? NBC некоторых ABC-переносчиков гидролизует АТФ, и эти переносчики используют энергию гидролиза для транспорта метаболитов через мембрану. Напротив, воротный механизм CFTR, вероятно, регулируется аденилаткиназной активностью, которой обладает С-терминальный участок NBF. Этот фермент катализирует перенос фосфата от АТФ на АМФ с образованием АДФ.
Предложен механизм, согласно которому связывание АТФ может вызывать димеризацию NBS и открывать канал, а АДФ, образующийся при действии аденилаткиназы, вызывает диссоциацию NBS и закрытие канала. Мутация CFTRAPhe508 нарушает работу канала на участке NBF и препятствует экспрессии его функционально полноценной формы.
Активность CFTR может регулироваться с участием других механизмов. Например, может происходить фосфорилирование регуляторного домена. Наряду с этим, при активации глутаматом может меняться селективность CFTR по отношению к разным анионам. Например, канал может начать пропускать хлорид-ионы и не пропускать НСO3-. Таким образом, CFTR, вероятно, функционирует как лиганд-зависимый анионный канал, а не как АТФ-зависимый ионный насос.
При кистозном фиброзе генетический дефект CFTR каналов приводит к нарушению трансэпителиального транспорта, что отражается на процессах секреции анионов и жидкостей в ряде органов. Согласно одной из гипотез, CFTR в основном работают как Cl--каналы, которые транспортируют хлорид в клетки, обеспечивая образование низкосолевой жидкости, увлажняющей воздухоносные пути, и ингибирование реабсорбции Na+ эпителиальными натриевыми каналами. Нарушение секреции через CFTR ионов Cl- при кистозном фиброзе приводит к поглощению натрия и воды эпителиальными клетками, что снижает объем жидкости, смачивающей воздухоносные пути, увеличивает вязкость и создает препятствия для ее очистки.
В зависимости от места мутации в гене CFTR, транспорт анионов может нарушаться в различной степени. Наряду с инфекциями воздухоносных путей, описанными выше, у больных может наблюдаться недостаточная секреция пищеварительных ферментов поджелудочной железы, непроходимость кишечника, бесплодие и усиленная потеря солей через потовые железы. Большинство больных рождаются с недостаточностью поджелудочной железы, приводящей к расстройствам пищеварения и к диарее.
Нарушение секреции бикарбоната и жидкости, вызванное мутациями в канале CFTR, приводит к потере функции поджелудочной железы и к прогрессивной деструкции ее ткани. Исследования, проводящиеся в настоящее время, направлены на возможность применения адресных препаратов для лечения дефектных функций CFTR или ENaC и на коррекцию процессов эпителиального транспорта у больных кистозным фиброзом.