Начиная с 1960-х годов, муковисцидоз — одно из наиболее известных моногенных заболеваний человека. Это самое частое фатальное аутосомно-рецессивное генетическое заболевание детей в европеоидных популяциях, со встречаемостью приблизительно 1 на 2500 родов и частотой носительства около 1 на 25. Позиционное клонирование (см. главу 10) гена муковисцидоза (названного CFTR) в 1989 г. и выделение тремя годами ранее гена мышечной дистрофии Дюшенна стали первыми примерами возможностей молекулярно-генетических методов по идентификации генов болезней.
Вскоре после клонирования гена муковисцидоза с помощью физиологических исследований было показано, что белок, кодируемый геном CFTR, регулирует хлорный канал, располагающийся в апикальной мембране эпителиальных клеток.
Фенотипы муковисцидоза
Болезнь поражает легкие и экзокринную функцию поджелудочной железы, но главный диагностический признак — повышение концентраций хлоридов и натрия в поте (часто впервые замечаемое, когда родители целуют детей). У большинства пациентов с муковисцидозом диагноз может основываться на легочной или панкреатической симптоматике и повышении уровня хлоридов пота. Менее чем 2% пациентов имеют нормальную концентрацию хлоридов пота, несмотря на типичные клинические проявления; в этих случаях надо проводить молекулярный анализ, устанавливающий наличие мутации в гене CFTR.
Легочная патология при муковисцидозе развивается в результате избыточной секреции бронхиального секрета и повторной инфекции; первоначально ее описывают как хроническую обструктивную болезнь легких, переходящую в бронхоэктазию. Хотя интенсивное лечение легких продлевает жизнь, в конце концов, наступает смерть от инфекции и легочной недостаточности. В настоящее время около половины пациентов доживают до 33 лет при очень вариабельном клиническом течении.
Нарушения функции поджелудочной железы при муковисцидозе — синдром мальабсорбции из-за недостаточной секреции панкреатических ферментов (липазы, трипсина, химотрипсина). Нормальное пищеварение и питание в основном могут восстанавливаться при приеме ферментов поджелудочной железы. От 5 до 10% пациентов с муковисцидозом имеют некоторую остаточную функцию поджелудочной железы для нормального пищеварения и называются панкреатически достаточными.
Пациенты с муковисцидозом с достаточной функцией поджелудочной железы лучше растут и имеют более благоприятный прогноз, чем большинство больных с недостаточностью. Клиническая гетерогенность патологии поджелудочной железы, по крайней мере частично, вызвана аллельной гетерогенностью, что обсуждается далее.
У больных муковисцидозом наблюдают множество различных фенотипов. Например, у 10-20% новорожденных с муковисцидозом после рождения встречается низкая кишечная непроходимость (меконеальный илеус), наличие которой требует исключить диагноз муковисцидоза. Затрагивается также половой тракт. Хотя женщины с муковисцидозом имеют лишь незначительное снижение фертильности, более 95% мужчин с муковисцидозом бесплодны, поскольку не имеют семявыносящих протоков, фенотип, известный как врожденная двусторонняя атрезия семявыносящих протоков.
В поразительном примере аллельной гетерогенности, вызывающей частичный фенотип, было обнаружено, что некоторые бесплодные мужчины, в остальном здоровы (т.е. не имеют легочных или панкреатических проявлений), имеют врожденную двустороннюю атрезию семявыносящих протоков, сцепленную со специфическими мутантными аллелями в гене муковисцидоза. Аналогично некоторые больные идиопатическим хроническим панкреатитом имеют мутации в гене CFTR при отсутствии других клинических признаков муковисцидоза.
Ген и белок CFTR при муковисцидозе
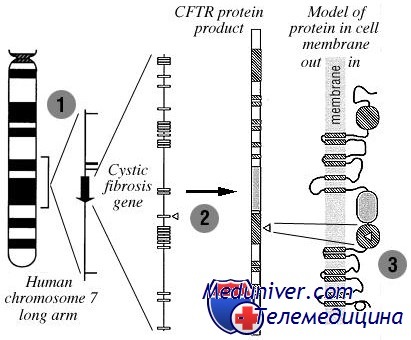
CFTR — ген в хромосоме 7q31, ассоциированный с муковисцидозом, содержит около 190 килобаз ДНК; кодирующая область с 27 экзонами; по предсказаниям, кодирует крупный трансмембранный белок размером около 170 килодальтон. На основе предсказанной функции белок, кодируемый CFTR, назван трансмембранным регулятором проводимости муковисцидоза (англ. Cystic Fibrosis Transmembrane conductance Regulator).
Его гипотетическая структура указывала, что белок должен принадлежать к так называемому семейству транспортных белков ABC (АТФ-связанных). По крайней мере 18 транспортных белков этого семейства вовлечены в развитие менделирующих и комплексных заболеваний.
Хлорный канал CFTR имеет пять областей: две области, связанные с прикреплением к мембране, каждая с шестью трансмембранными последовательностями; две области связи с АТФ; и регуляторная область с многочисленными сайтами фосфорилирования. Значение каждой области доказано идентификацией в каждой из них вызывающих муковисцидоз миссенс-мутаций.
Отверстие хлорного канала формируется 12 трансмембранными сегментами. АТФ связывается и гидролизуется в нуклеотидной области, полученная энергия используется для открывания и закрывания канала. Управление канала связано, по крайней мере частично, с фосфорилированием регуляторного домена.
Патофизиология муковисцидоза
Муковисцидоз — следствие аномального транспорта жидкостей и электролитов через апикальные мембраны эпителия. Эта аномалия приводит к патологии легких, поджелудочной железы, кишечника, гепатобилиарного дерева и мужского полового тракта. Патофизиологические аномалии наиболее хорошо объяснены для потовых желез.
Снижение функции CFTR означает, что хлориды не могут реабсорироваться в канале потовых желез, приводя к уменьшению электрохимического градиента, в норме управляющего движением натрия через апикальную мембрану. Этот дефект, в свою очередь, ведет к повышению концентрации хлоридов и натрия в поте. Влияние на транспорт электролитов аномалий в белке CFTR также тщательно изучено в дыхательных путях и эпителии поджелудочной железы.
В легких повышенное поглощение натрия и сниженная секреция хлоридов приводят к уменьшению поверхностной жидкости дыхательных путей. Следовательно, слой слизи может прилипать к поверхности клеток, нарушая откашливание и отхождение слизи, обеспечивая благоприятные условия для синегнойной палочки (Pseudomonas aeruginosa), основного возбудителя хронической легочной инфекции при муковисцидозе.
Генетика муковисцидоза
Мутации в полипептиде CFTR при муковисцидозе. Первая идентифицированная мутация при муковисцидозе, делеция остатка фенилаланина в позиции 508 (F508), в первой области, связывающей АТФ (NBD1), — самый частый дефект, составляющий до 70% всех аллелей муковисцидоза в европеоидных популяциях. В этих популяциях только семь других мутаций встречаются с частотой выше 0,5%. Описаны все типы мутаций, но наибольшая группа (почти половина) — миссенс-замены.
Остальные представляют точковые мутации других типов, менее 1% — геномные перегруппировки. Хотя выявлено более 1200 связанных с болезнью вариантов последовательности гена муковисцидоза, фактическое количество патогенных миссенс-мутаций отчасти остается неопределенным, поскольку не все подвергнуты функциональному анализу.
Хотя биохимические аномалии, связываемые с большинством мутаций при муковисцидозе, неизвестны, описаны четыре общих механизма нарушения белковой функции. Мутации 1-го класса вызывают нарушения в синтезе белка, например связанные с преждевременными стоп-кодонами или мутациями, приводящими к нестабильности РНК. Поскольку CFTR — гликозилированный трансмембранный белок, он должен обрабатываться и гликозилироваться в эндоплазматическом ретикулуме и комплексе Гольджи; мутации 2-го класса — результат дефекта белка, вызывающего нарушение его третичной структуры.
Этот класс иллюстрирует мутация F508, мутантный белок нормально не складывается и не может выйти из эндоплазматического ретикулума. Тем не менее фенотип белка F508 комплексный: кроме нарушения складывания, белок также имеет дефекты в устойчивости и активизации.
Существенные функции нуклеотид-сцепленных областей и регуляторной области иллюстрируются случаем вызывающих муковисцидоз мутаций, нарушающих регулирование белка (мутации 3-го класса). Мутации 4-го класса располагаются в мембранной области и, соответственно этой локализации, приводят к нарушению проведения хлоридов. Мутации 5-го класса уменьшают число копий CFTR. Мутантные белки класса 6 синтезируются нормально, но неустойчивы на поверхности клетки.
Генокопирование при муковисцидозе: мутации в гене эпителиального натриевого канала SCNN1
Хотя CFTR — единственный ген, сцепленный с классическим муковисцидозом, обнаружено несколько семей с неклассическими проявлениями (включая муковисцидоз-подобные легочные инфекции с менее тяжелыми нарушениями пищеварения и повышением уровня хлоридов пота), имеющих мутации в гене эпителиального натриевого канала SCNN1.
Это соответствует функциональному взаимодействию белка CFTR и эпителиального канала натрия. Основное его клиническое значение в настоящее время — демонстрация того, что пациенты с неклассическим муковисцидозом могут иметь локусную гетерогенность, и если мутации в гене CFTR не обнаружены, нужно искать аномалии в гене SCNN1.
Корреляции генотипа и фенотипа при муковисцидозе. Поскольку все пациенты с классической формой муковисцидоза имеют мутации в гене муковисцидоза, клиническая гетерогенность при муковисцидозе обусловлена аллельной гетерогенностью, эффектами других модифицирующих локусов или негенетических факторов. Из генетического и клинического анализа пациентов с муковисцидозом возникли два обобщения.
Во-первых, генотип CFTR дает возможность точного прогноза экзокринной функции поджелудочной железы. Например, пациенты, гомозиготные по частой мутации F508 или другим аллелям с нарушенным синтезом белка (например, преждевременным стоп-кодонам), обычно имеют недостаточность поджелудочной железы. С другой стороны, аллели, допускающие синтез частично функционального белка CFTR, например Argll7His, обычно имеют достаточную функцию поджелудочной железы. Во-вторых, генотип CFTR не дает оснований для прогнозирования тяжести патологии легких.
Например, тяжесть патологии легких варьирует среди гомозиготных по мутации F508 пациентов. Причины такой слабой корреляции генотипа и фенотипа для патологии легких непонятны. Недавно было сообщение о выявлении гена-модификатора патологии легких при муковисцидозе, гене, кодирующем TGFbl. Два варианта TGFbl ассоциируются с более тяжелой патологией легких при муковисцидозе. Если данный факт окажется достоверным, это может обеспечить понимание патологических механизмов, лежащих в основе патологии легких и расширить терапевтические возможности.