Болезнь Кауден — редкий наследственный синдром с аутосомно-доминантным путем передачи (локус гена неизвестен), получивший свое название по фамилии семьи, в которой впервые были выявлены такие больные. Причина неизвестна. Не обнаружено нарушений в хромосомном наборе, репарации ДНК, повышения частоты поломки хромосом в культуре лимфоцитов. Вместе с тем отмечен дефект регуляции клеточной пролиферации.
Болезнь Кауден проявляется в возрасте от 4 до 75 лет, в половине случаев — до 40 лет. Мужчины болеют чаще. Характеризуется множественными тамартомами (нейроэктодермального, мезодермального и эктодермального происхождения) в ассоциации со злокачественными опухолями молочной (нередко двусторонними), щитовидной желез, желудочно-кишечного тракта (пищевод, двенадцатиперстная и ободочная кишка и т.д.), предстательной железы, матки. При болезни Кауден также обнаруживаются кистозно-фиброзная мастопатия с ранней злокачественной трансформацией (у мужчин — гинекомастия), зоб, полипоз желудочно-кишечного тракта. Поражение центральной нервной системы проявляется умственной отсталостью, эпилепсией, такими опухолями, как невриномы, ганглио невромы, менинтиомы. Могут наблюдаться нарушения менструального цикла, нарушения опорно-двигательной системы (увеличение размера черепа, кифосколиоз, готическое нёбо).
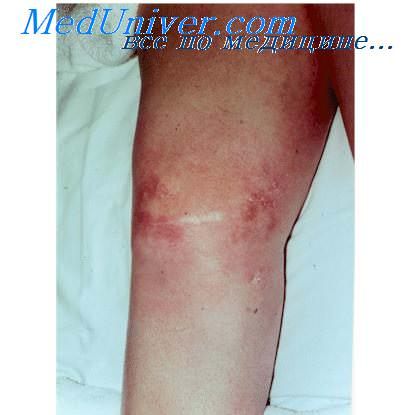
Кожные проявления болезни Кауден представлены преимущественно (89%) трихилеммомами, а также другими доброкачественными опухолями придатков кожи. Кроме того, при болезни Кауден могут возникать папилломы десен и веррукозная гиперплазия десен, папилломатоз губ, акральный кератоз (напоминающий акрокератоз Хопфа), базалиомы, плоскоклеточный рак кожи, меркелиома, меланома, липома, ангиома.
Трихолеммомы болезни Кауден имеют вид мелких папул, похожих на плоские бородавки, цвет которых может не отличаться от окружающей здоровой кожи, быть розовым или коричневым. Они локализуются в центральной части лица, вокруг рта, на губах, ушных раковинах. Иногда множественность трихолеммом уродует больного. Они обычно предшествуют развитию рака внутренних органов.
Течение болезни Кауден сопровождается возникновением новых опухолей на протяжении всей жизни.
Диагноз болезни Кауден устанавливается на основании анамнеза, клинической картины, данных лучевой диагностики (маммография, ультразвуковое исследование щитовидной железы), выявления трихолеммом при гистологическом исследовании. Важна ранняя диагностка трихолеммом как возможного маркера повышенного риска развития рака молочной железы (20%).
Дифференциальный диагноз болезни Кауден проводится с множественными сиринтомами, множественными трихоэпителиомами, плоскими бородавками, синдромом Мюир—Торре, гиалинозом кожи и слизистых оболочек.
При лечении опухолей кожи болезни Кауден используют лучи лазера или электрокоагуляцию. Необходимо медико-генетическое консультирование, а также наблюдение у онколога с целью исключения рака молочной и щитовидной желез. Высокий риск развития рака молочной железы иногда требует проведения профилактической двухсторонней мастэктомии.
Болезнь Кауден
Синдром Мюир—Торре
Синдром Мюир—Торре — редкое заболевание с аутосомно-доминантным типом наследования, характеризующееся сочетанием опухолей кожи и множественных новообразований внутренних органов. Проявляется на 5-6-м десятилетиях жизни. Генетическая основа заболевания неясна. Предполагается, что оно является вариантом семейного ракового синдрома Линча. Потомки родственников с синдромом Линча страдали синдромом Мюир—Торре, у них отмечена связь заболевания с измениями в хромосоме 2(р).
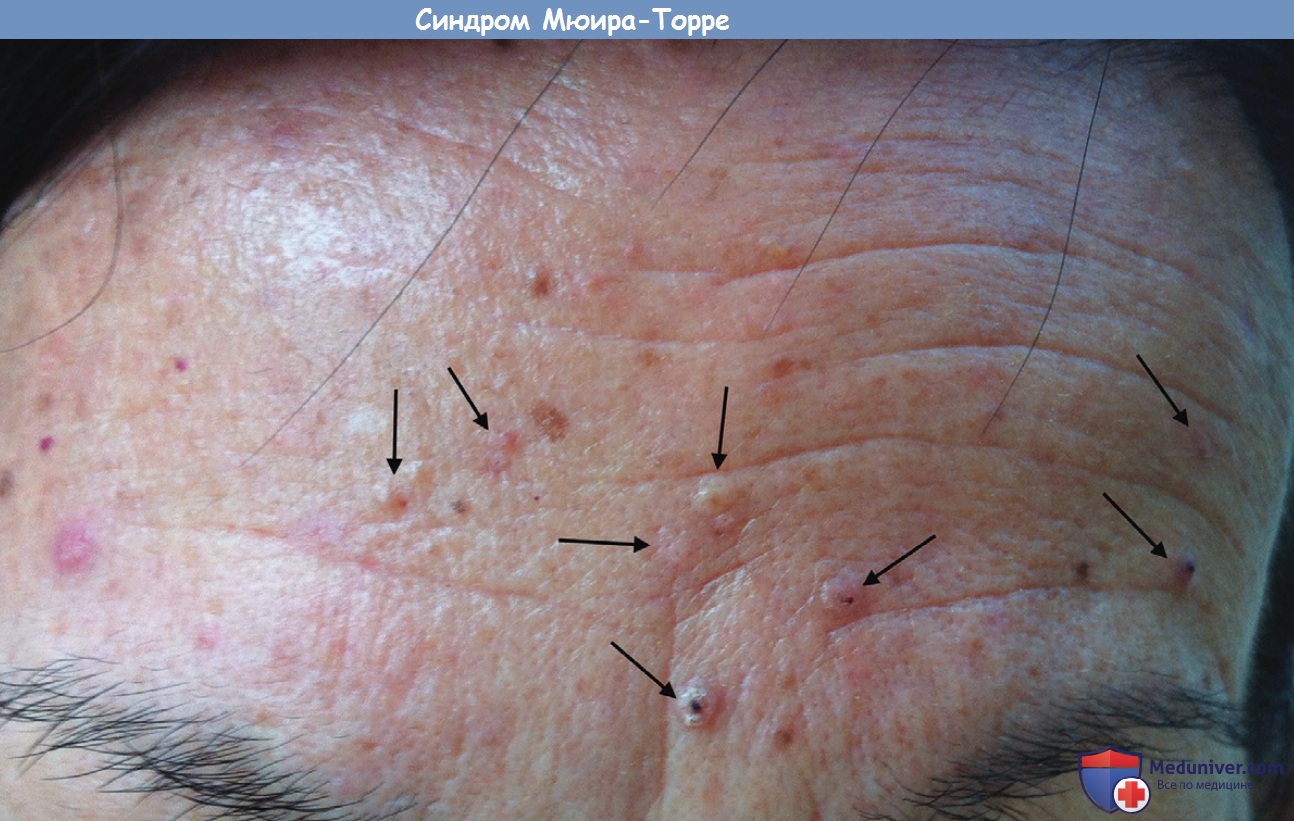
Среди кожных опухолей синдрома Мюир-Торре помимо множественных аденом и раков сальных желез встречаются кератоакантомы, солнечный кератоз, базалиомы с сально-железистой дифференци-ровкой, меланомы. В отдельных случаях встречаются солитарные аденомы сальных желез. Кожные опухоли обычно небольших размеров (не более 1 см в диаметре), чаще располагаются на лице, примерно в половине случаев они возникают после развития злокачественных новообразований внутренних органов. Наиболее частыми из новообразований внутренних органов являются развившиеся на фоне полипов аденокарциномы толстой кишки. Также часто встречается рак желудка, тошей. двенадцатиперстной кишки, мочевого пузыря, почки, предстательной железы, эндометрия, вульвы, яичников, гортани, легких. Новообразования внутренних органов при синдроме Мюир—Торре отличаются низкой степенью злокачественности, поэтому продолжительность жизни таких больных выше, чем больных с аналогичной локализацией рака без синдрома Мюир—Торре.
Диагноз синдрома Мюир-Торре основывается на сочетании кожных новообразований и опухолей внутренних органов. Примерно в 30% случаев кожные опухоли являются первым симптомом заболевания, что способствует раннему выявлению рака внутренних органов.
Дифференциальный диагноз синдрома Мюир-Торре проводится с ненаследственными опухолями кожи и внутренних органов.
При лечении опухолей кожи синдрома Мюир-Торре эффективны ароматические ретиноиды. Профилактика заключается в медико-генетическом консультировании и обследовании членов семьи у онколога, а также в ограничении воздействия на организм канцерогенных веществ.
Множественная эндокринная неоплазия III типа
Множественная эндокринная неоплазия III типа (син.: множественная эндокринная неоплазия lib типа, синдром множественной слизистой невромы) обычно наследуется по аутосомно-доминантному типу, хотя до 50% случаев могут быть спорадическими. У 93-95% больных обнаруживают точечные мутации протоонкогена c-ret. Этот протоонкоген кодирует рецептор нейротропного фактора, регулирующего пролиферацию и дифференцировку клеток — производных неврального гребешка. Мутации c-ret приводят к активации рецептора тирозинкиназы и трансформации нейроэктодермальных клеток.
Три главных компонента синдрома множественной эндокринной неоплазии включают множественные невромы слизистых оболочек, медуллярный рак щитовидной железы (95%), феохромоцитому (33%), однако только 50% больных имеет полную триаду.
Элементы синдрома множественной эндокринной неоплазии на слизистых оболочках отмечаются у 95% больных, они возникают в начале жизни, обычно задолго до злокачественных новообразований внутренних органов, что имеет большое диагностическое значение. Определяются мелкие, гладкие узлы, преимущественно на губах, языке щеках, придающие слизистой оболочке бугристый вид, иногда поражаются веки, конъюнктива и роговица. Часто встречаются следующие проявления: марфаноподобная внешность, признаки прогерии, воронкообразная грудь, утолщение нервов роговицы, гантлионевромы желудочно-кишечного тракта, мегаколон.
Диагноз синдрома множественной эндокринной неоплазии устанавливается клинически. Ранние изменения нервов можно обнаружить при гистологическом исследовании биоптатов кожи.
Течение синдрома множественной эндокринной неоплазии. Заболевание прогрессирует медленно. Однако медуллярный рак щитовидной железы и феохромоцитома при этом синдроме характеризуются быстрым ростом и выраженной склонностью к метастазированию.
План лечения синдрома множественной эндокринной неоплазии зависит от всех компонентов синдрома. Например, если одновременно имеются рак щитовидной железы и феохромоцитома, сначала проводят операцию на надпочечниках, чтобы предотвратить катехоламиновый криз во время операции на щитовидной железе.
Расшифровка генетических дефектов, лежащих в основе синдрома множественной эндокринной неоплазии, позволяет оценить риск заболевания у родственников больных и планировать профилактические мероприятия.