Диагностика гипергонадотропного гипогонадизма затруднена до наступления полового созревания. У большинства пациенток с данным нарушением клинические проявления отсутствуют до наступления половой зрелости (исключение — синдром Тернера).
а) Синдром Тернера. Тернер описал синдром, включающий половой инфантилизм, крыловидную шею и вальгусную деформацию локтевого сустава у взрослых женщин (глава 98). Ульрих описал женщину невысокого роста, обладающую многими из описанных выше фенотипических особенностей. Термин «синдром Ульриха-Тернера» часто используется в Европе, но редко используется в США, где это состояние называется синдромом Тернера. Этот синдром определяется как сочетание характерных фенотипических признаков, сопровождающееся полным или частичным отсутствием второй Х-хромосомы с мозаицизмом или без него.
1. Патогенез. У половины пациенток с синдромом Тернера — хромосомный набор 45,X. У 15% пациенток наблюдается мозаичная форма синдрома, представленная 45,X и нормальной клеточной линей (45,Х/46,ХХ). Др. мозаичные формы с изохромосомами, 45,X/46,X,i(Xq); с кольцевыми хромосомами, 45,Х/46,Х,r(Х)-, или с фрагментами хромосом, 45,X/46fra, встречаются реже. Мозаицизм обнаруживается при исследовании образцов более одной ткани. У 80% пациенток с кариотипом 45,X единственная Х-хромосома имеет материнское происхождение. Механизм потери хромосом неизвестен, и риск развития синдрома не увеличивается с возрастом матери. Гены, участвующие в характерном для синдрома Тернера фенотипе, связаны с Х-хромосомой, они избегают инактивации.
Основной локус, участвующий в контроле линейного роста, локализуется в псевдоавтосомной области Х-хромосомы (PAR1). SHOX — гомеобокс-содержащий ген размером 170 тпн ДНК, расположенный в PAR1, который важен для контроля роста у детей с синдромом Тернера, синдромом Лери-Вейлля и (реже) у пациентов с идиопатической низкорослостью. Предполагается, что гены, контролирующие нормальную функцию яичников, находятся на Хр, а два супергена — на Xq.
Синдром Тернера встречается у 1:500-2500 живорожденных девочек. Во время зачатия частота кариотипа 45,X составляет ~3%, но при этом в 99% случаев беременность спонтанно прерывается (5-10% от общего числа самопроизвольных выкидышей). Мозаицизм (45,X/46,XX) встречается с большей частотой, чем при любом др. анеуплоидном состоянии, однако мозаичная форма синдрома Тернера редко встречается среди абортированных эмбрионов; эти данные указывают на преимущественное выживание плодов с мозаичными формами синдрома.
В норме яичник плода содержит 7 млн ооцитов, при этом они начинают быстро исчезать после 5 мес беременности. При рождении их насчитывается всего 2 млн (1 млн активных фолликулов); при наступлении менархе — 400 000-500 000; а ко времени наступления менопаузы остается 10 000. При отсутствии одной Х-хромосомы этот процесс ускоряется, и почти все ооциты исчезают к 2-летнему возрасту. У абортированных плодов с кариотипом 45,X количество первичных половых клеток в половом гребне находится в пределах нормы. Это позволяет предположить, что у пациенток с синдромом Тернера ускоряется нормальный процесс потери ооцитов. В конечном итоге яичники формируются в виде тяжей и состоят только из соединительной ткани с малым количеством половых клеток.
2. Клинические проявления. Многих пациенток с синдромом Тернера можно распознать при рождении из-за характерного отека тыльной стороны кистей и стоп и рыхлых кожных складок у основания шеи. Часто отмечаются НМТ при рождении и уменьшение длины тела. К клиническим проявлениям в детском возрасте относятся перепончатая шея (рыхлые складки кожи на шее), низкое расположение линии роста волос на затылке, маленькая нижняя челюсть, оттопыренные уши, эпикантальные складки, высокое куполообразное нёбо, широкая ГК, создающая иллюзию широко расставленных сосков, вальгусная деформация локтевого сустава и выпуклые ногти. Часто предполагаемый диагноз впервые ставится в период полового созревания, когда не происходит развития МЖ.
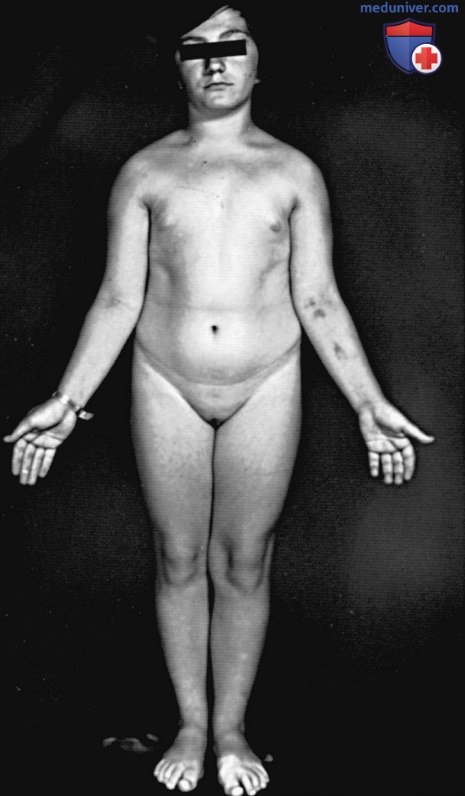
Невысокий рост — основной симптом практически у всех женщин с синдромом Тернера — может присутствовать наряду с относительно небольшим количеством др. клинических проявлений. Замедление скорости линейного роста начинается в младенчестве и раннем детстве, постепенно становится более выраженным в позднем детстве и подростковом возрасте, приводит к значительной низкорослости у взрослого человека. Половое созревание (развитие МЖ) в ожидаемом возрасте не происходит, но наблюдаются признаки адренархе (лобковые волосы). Средний рост взрослого человека среди пациенток с синдромом Тернера, не получавших лечения, в США и большей части Северной Европы составляет 143-144 см, в Аргентине — 140 см, а в Скандинавии — 147 см (рис. 1). Рост ребенка хорошо коррелирует со средним ростом родителей (средний рост родителей с поправкой на пол ребенка). Для оценки роста у женщин с синдромом Тернера разработаны специальные кривые роста.
Рисунок 1. Синдром Тернера у девушки 15 лет с нарушением полового созревания, низким ростом, вальгусной деформацией локтевого сустава и зобом. Крыловидная шея отсутствует. Кариотипирование выявило хромосомный набор 45,Х/46,ХХ
Часто встречаются сопутствующие пороки сердца. Опасные для жизни последствия гаплонедостаточности Х-хромосомы у женщин с синдромом Тернера затрагивают ССС. У взрослых с синдромом Тернера отмечается 4-5-кратное увеличение преждевременной смертности, вторичной по отношению к ВПС и преждевременному развитию ИБС. У пациенток с синдромом Тернера встречаются бессимптомные пороки сердца: напр., двустворчатый аортальный клапан, а также расширение восходящего отдела аорты, коарктация аорты и частичное аномальное впадение легочных вен. Независимо от возраста на момент постановки диагноза, все пациентки с синдромом Тернера нуждаются в комплексном обследовании ССС у кардиолога, специализирующегося на ВПС. Полное кардиологическое обследование, включающее ЭхоКГ, приводит к обнаружению изолированных нестенозирующих двустворчатых аортальных клапанов у 1/3-1/2 пациентов.
В более позднем возрасте болезнь двустворчатого аортального клапана может прогрессировать, приводя к расширению корня аорты. Более редкие пороки: коарктация аорты (20%), стеноз аортального клапана, ПМК и аномальное впадение легочных вен. В одном исследовании пороки развития ССС наблюдались у 38% пациенток с хромосомным набором 45,X по сравнению с 11% пациенток с мозаичной формой моносомии Х-хромосомы. Наиболее распространенными были пороки аортального клапана и коарктация аорты. Крыловидная шея у пациенток с диагностированными хромосомными синдромами и без них ассоциирована с пороками сердца, связанными и не связанными с нарушением тока крови. Среди всех пациенток с синдромом Тернера у лиц с крыловидной шеей шансы выявления коарктации аорты гораздо выше, чем у лиц без этой патологии.
Трансторакальная ЭхоКГ у молодых женщин обеспечивает адекватный объем обследования, если четко визуализируются анатомические особенности сердца. В противном случае у бессимптомных пациенток с синдромом Тернера в качестве скринингового исследования следует рассматривать магнитно-резонансную ангиографию. В подростковом возрасте и до наступления беременности (если она возможна) следует рассмотреть возможность повторного обследования сердца даже у лиц с не выявленными ранее аномалиями ССС. Следует регулярно контролировать АД даже при отсутствии поражений сердца или почек, особенно у пациенток с признаками расширения корня аорты. МРТ сердца — ценный инструмент для выявления и мониторинга расширения корня аорты.
Всем женщинам с синдромом Тернера при постановке диагноза следует проводить УЗИ почек. У 1/4-1/3 пациенток при УЗИ выявляются пороки развития почек (50% из них при кариотипе 45,X). К более серьезным нарушениям относятся тазовая почка, подковообразная почка, удвоение чашечно-лоханочной системы, полное отсутствие одной почки и непроходимость лоханочно-мочеточникового сегмента. Некоторые пороки развития могут увеличивать риск развития гипертонии и ИМП. Часто встречается идиопатическая гипертония. У женщин с синдромом Тернера и нормальными результатами исходного УЗИ почек не отмечалось развития почечной недостаточности в течение периода наблюдения, составлявшего в среднем 6 лет.
При обследовании яичников с помощью УЗИ более ранние исследования показали снижение процента обнаружения яичников с младенчества до позднего детства. В последующем докладе при поперечном и продольном исследовании не было выявлено таких возрастных различий. У 27-46% пациенток яичники обнаруживались в различном возрасте; также яичники обнаруживались у 76% пациенток с мозаицизмом по Х-хромосоме и у 26% пациенток с кариотипом 45,X.
Обычно половое созревание не наступает, но у 10-20% женщин отмечается спонтанное развитие МЖ, а у небольшого процента могут наблюдаться менструации. Первичная недостаточность половых желез ассоциирована с ранним началом адренархе (повышения уровня дегидроэпиандростерона-сульфата), но задержкой пубархе (роста волос на лобке). У менструирующих пациенток с синдромом Тернера отмечены случаи спонтанной беременности. Поступали сообщения о преждевременной менопаузе, повышенном риске выкидыша и потомстве с повышенным риском трисомии 21. У женщины с кариотипом 45,Х/46,Х,r(Х), получавшей заместительную гормональную терапию, было три беременности, закончившихся рождением ребенка мужского пола с нормальным кариотипом 46,XX, самопроизвольным выкидышем и рождением здорового доношенного ребенка женского пола с синдромом Тернера и кариотипом 45,Х/46,Xr(Х).
Антитиреоидные АТл (АТл к тиреоидной пероксидазе или к тиреоглобулину) встречаются у 30-50% пациенток. Распространенность с возрастом увеличивается. Аутоиммунные заболевания ЩЖ с развитием зоба или без него встречаются у 10-30% пациенток. У пожилых пациенток с синдромом Тернера наблюдаются возрастные нарушения углеводного обмена, характеризующиеся нарушением толерантности к глюкозе и инсулинорезистентностью, и редко — СД-2. У женщин с кариотипом 45,X описано нарушение секреции инсулина. В подростковом возрасте у пациенток отмечается повышенный уровень холестерина независимо от ИМТ или кариотипа.
Сообщалось о развитии ВЗК (болезни Крона и язвенного колита), ЖКК из-за аномалий брыжеечной сосудистой сети и замедлении эвакуации содержимого желудка. Поскольку при синдроме Тернера повышается риск развития глютеновой энтеропатии, при этом заболевание поражает 4-6% пациентов, рекомендуется проводить скрининг на наличие глютеновой энтеропатии. Несмотря на связь синдрома Тернера и аутоиммунных заболеваний, распространенность СД-1 у лиц с синдромом Тернера не очень высока.
Пороки развития грудины м.б. обнаружены с помощью РОГК в боковой проекции. Увеличение угла между плечом и предплечьем не имеет клинического значения. У 10% девочек-подростков встречается сколиоз. Врожденная дисплазия ТБС встречается чаще, чем в общей популяции. Зрительные нарушения включают дисгенезию переднего сегмента глаза и кератоконус. Пигментные невусы становятся более заметными с возрастом; часто встречаются меланоцитарные невусы. Первичный гипергидроз, нижнечелюстной валик и гнездная алопеция встречаются редко.
Рецидивирующий двусторонний средний отит развивается у ~75% больных. Часто встречаются нейросенсорные нарушения слуха, и их распространенность увеличивается с возрастом. Проблемы с сенсомоторной интеграцией грубой и тонкой моторики, неспособность ходить до 15 мес и ранняя речевая дисфункция часто вызывают вопросы о предполагаемой задержке развития, но интеллект у большинства пациентов нормальный. У пациентов с кариотипом 45,Х/46,Х,г(Х) действительно возникают когнитивные нарушения; кольцевая хромосома не инактивируется и приводит к появлению двух функциональных Х-хромосом.
Особое внимание у женщин с синдромом Тернера следует уделять психосоциальному развитию. У женщин с синдромом Тернера поведение нормальное, но они подвержены повышенному риску социальной изоляции, инфантильности и тревожности. У женщин с синдромом Тернера зарегистрированы др. нарушения: дислексия, невербальное расстройство обучения и СДВГ. У взрослых нарушение способности к пространственному восприятию встречается чаще, чем в общей популяции. Некоторые неподтвержденные данные предполагают существование импринтированного локуса, сцепленного с Х-хромосомой, который влияет на когнитивные функции — вербальные навыки и навыки самоорганизации высшего порядка. Данные функции выражены лучше, когда Х-хромосома имеет отцовское происхождение.
Распространенность мозаицизма во многом зависит от методов, используемых для изучения хромосомных паттернов. Использование флюоресцентной гибридизации in situ и ПЦР с обратной транскрипцией увеличило распространенность мозаичных паттернов до 60-74%.
Мозаицизм с участием У-хромосомы встречается в 5% случаев. Популяционное исследование с использованием ПЦР с пятью различными наборами праймеров обнаружило хромосомный материал У-хромосомы в 12,2% случаев. Гонадобластома у пациенток с У-хромосомой встречалась в 7-10% случаев. Поэтому рекомендуется проводить профилактическую гонадэктомию даже при отсутствии признаков опухолей на МРТ или КТ. Лучшее время для проведения данной процедуры — период постановки диагноза, но в будущем может потребоваться пересмотр этой практической рекомендации. Локус гонадобластомы на У-хромосоме (GBY) локализуется вблизи У-центромеры. Наличия только локуса SRY (определяющего пол участка У-хромосомы) недостаточно для обеспечения повышенной восприимчивости к развитию гонадобластомы.
Детальное обследование 53 пациенток с синдромом Тернера методом вложенной ПЦР практически во всех случаях исключило низкоуровневый У-мозаицизм. Второй раунд ПЦР выявил SRY на дистальном участке короткого плеча У-хромосомы только у двух испытуемых. Поэтому рутинное проведение ПЦР для выявления У-хромосомы и определения риска развития гонадобластомы не показано. Высокопроизводительное генотипирование — эффективный и недорогой метод идентификации аномалий Х-хромосомы и выявления материала У-хромосомы.
У пациенток с мозаицизмом 45,X/46,XX клинических нарушений меньше и они менее выражены. Низкий рост встречается так же часто, как и у пациенток с кариотипом 45,X, и м.б. единственным проявлением данного состояния, кроме недостаточности яичников (см. рис. 1).
3. Лабораторные признаки. У пациенток с низкорослостью следует рассмотреть рутинное применение хромосомного анализа. При систематическом поиске с использованием анализа ДНК лейкоцитов методом сазерн-блоттинга синдром Тернера был выявлен у 4,8% женщин, обратившихся за консультацией эндокринолога по причине невысокого роста. У пациенток с маркерной хромосомой в некоторых или во всех клетках следует определить последовательность ДНК в центромере У-хромосомы или вблизи нее с целью выявления локуса GBY.
После установления диагноза показаны УЗИ сердца, почек и яичников. Наиболее распространенные скелетные аномалии — укорочение 4-й плюсневой и пястной костей, эпифизарный дисгенез в коленных и локтевых суставах, деформация Маделунга (порок развития лучезапястного сустава, клинически проявляющийся выстоянием головки локтевой кости к тылу и смещением кисти в ладонную сторону), сколиоз и недостаточная минерализация костей у пожилых пациенток.
Уровни гонадотропинов (особенно ФСГ) в плазме крови заметно повышены, достигают в младенчестве более высоких значений, чем в контрольной группе, сопоставленной по возрасту. В 2-3 года происходит прогрессирующее снижение уровней гормонов, которые достигают низшего уровня в 6-8 лет, а к 10-11 годам поднимаются до уровня взрослых пациенток, перенесших процедуру кастрации.
Следует проводить периодическое обследование на наличие АТл к тиреоидной пероксидазе и тиреоглобулину для выявления аутоиммунного тиреоидита. В случае «+» результата следует исследовать уровни тироксина и ТТГ. Женщинам с синдромом Тернера следует провести скрининговое обследование на глютеновую энтеропатию путем измерения тканевых IgA к трансглутаминазе. Первичное обследование должно проводиться в 4 года и повторяться каждые 2-5 лет. Широкомасштабные исследования не смогли установить факт того, что дефицит СТГ играет основную роль в патогенезе нарушения роста. Нарушения в нормальном характере СТГ отмечаются у подростков из-за недостатка половых стероидов, но не выявлены у маленьких детей женского пола с синдромом Тернера. Моноциты и лимфоциты in vitro демонстрируют пониженную чувствительность к инсулиноподобному фактору роста 1.
4. Лечение. Лечение рекомбинантным человеческим гормоном роста увеличивает скорость роста и итоговый рост у большинства, но не у всех детей с синдромом Тернера. При раннем начале лечения многие женщины достигают роста >150 см. В крупном многоцентровом плацебо-контролируемом клиническом исследовании 99 пациентов с синдромом Тернера, которые начали получать гормон роста 10,9 года в дозировке от 0,27-0,36 мг/кг/нед, достигли среднего роста 149 см. При этом 1/3 достигла роста >152,4 см (60 дюймов). В Нидерландах применение более высоких доз гормона роста (до 0,63 мг/кг/нед в течение 3-го года лечения) привело к тому, что к взрослому возрасту 85% испытуемых достигли роста в нормальном диапазоне для голландской референтной группы населения. Лечение гормоном роста следует начинать в раннем детском возрасте или при наличии признаков снижения скорости роста на специфических кривых роста для синдрома Тернера. Терапия гормоном роста у пациенток с синдромом Тернера не приводит к существенному ухудшению толерантности к углеводам и выраженным побочным явлениям.
Если пациентка получает высокие дозы гормона роста, следует контролировать уровень инсулиноподобного фактора роста 1 в сыворотке крови. Если уровень инсулиноподобного фактора роста 1 значительно повышен, потребуется снижение дозы гормона роста. Лечение гормоном роста может привести к избыточному росту кистей и стоп у некоторых женщин с синдромом Тернера.
Также для лечения низкорослости, связанной с синдромом Тернера, используется оксандролон в качестве монотерапии либо в сочетании с гормоном роста. Этот синтетический анаболический стероид обладает слабым андрогенным эффектом. Во время терапии следует наблюдать пациенток на предмет признаков пубархе и гепатотоксичности (встречается редко).
Показана заместительная терапия эстрогенами, но единого мнения относительно оптимального возраста начала лечения не существует. Следует учитывать психологическую готовность пациентки к принятию лечения. Улучшение роста, достигнутое у девушек, получавших лечение гормоном роста в детстве, позволяет начать заместительную терапию эстрогенами в 12-13 лет. Отсрочка терапии эстрогенами до 15 лет для оптимизации потенциала роста кажется неоправданной. Изменение в пользу более раннего начала терапии эстрогенами обусловлено психологической важностью полового созревания в соответствии с возрастом. Отсроченное лечение эстрогенами может пагубно влиять на здоровье костей и др. аспекты здоровья ребенка.
Заместительная терапия низкими дозами эстрогенов в 12 лет обеспечивает нормальный темп полового созревания, не препятствуя «+» влиянию гормона роста на конечный рост взрослого человека. Терапия эстрогенами улучшает вербальную и невербальную память у женщин с синдромом Тернера. Молодые женщины, у которых половое развитие соответствует возрасту, достигшие нормального роста, продемонстрировали нормальные результаты при заполнении опросников по качеству жизни, связанному со здоровьем.
Существует много форм эстрогенных ЛП. Ранее использовались эстрогены для перорального применения. Большую популярность набирают трансдермальные пластыри: при использовании действующее в-во не проходит через печень, и для достижения адекватного лечебного эффекта требуется лишь небольшое количество эстрогена. К пероральным ЛП, эффективно индуцирующим половое созревание, относятся эстрогены конъюгированные (Премарин) в ежедневной дозе 0,15-0,625 мг или микронизированный эстрадиол (эстрейс), назначаемый в ежедневной дозе 0,5 мг 3-6 мес. Рекомендуемые дозы при применении трансдермального пластыря составляют 6,25 мкг/сут с постепенным увеличением дозы в течение 2 лет до взрослой дозы в 100-200 мкг/сут. ЛП эстрогена можно принимать циклично (в дни 1-23 цикла) или непрерывно.
Дополнительно следует назначить гестагенный ЛП медроксипрогестерон («Провера») (принимается на 10-23-й сутки цикла) в ежедневной дозе 5-10 мг. Через неделю после приема гестагена возникает кровотечение отмены. КОК также могут использоваться для заместительной гормональной терапии.
Пренатальный хромосомный анализ, показанный по причине позднего репродуктивного возраста, выявлял кариотип 45,X/46,XX с частотой, в 10 раз превышающей частоту при постнатальной диагностике. У большинства этих пациенток клинические проявления синдрома Тернера отсутствуют, а уровень гонадотропных гормонов находится в норме. Осведомленность о существовании этого незначительно выраженного фенотипа важна при консультировании пациенток.
Психосоциальная поддержка этих женщин считается неотъемлемым компонентом лечения. Всестороннее психопедагогическое обследование рекомендуется проводить либо в момент постановки диагноза синдрома Тернера, в зависимости от возраста пациента, когда становится очевидным какой-либо из компонентов поведения или когнитивной деятельности, либо непосредственно перед поступлением в школу. Общество синдрома Тернера с местными отделения в США и аналогичными группами в Канаде и др. странах обеспечивает ценную систему поддержки этих пациентов и их семей в дополнение к поддержке, предоставляемой командой медработников.
При использовании донорства яйцеклеток и ЭКО удалось добиться успешных доношенных беременностей. У подростков с незначительными признаками спонтанного полового созревания отмечены яичники с фолликулами. Это делает возможным использование криоконсервированной ткани яичников с незрелыми яйцеклетками до наступления обратного развития яичников для достижения беременности в будущем. У взрослых женщин с синдромом Тернера высока распространенность недиагностированных нарушений минеральной плотности костной ткани, липидного обмена и функции ЩЖ. Часто встречаются нарушение толерантности к глюкозе со снижением первой фазы реактивной секреции инсулина, повышенное АД и снижение безжировой массы тела. При заместительной терапии ЛП половых гормонов толерантность к глюкозе ухудшается, но безжировая масса тела, АД и общее физическое развитие улучшаются. Нейрокогнитивный профиль взрослых женщин не зависит от эстрогенного статуса.
б) ХХ-дисгенезия гонад. В некоторых случаях у фенотипически и генетически нормальных женщин могут отмечаться поражения половых желез, идентичные таковым у пациенток с кариотипом 45,X, но без соматических признаков синдрома Тернера. Их состояние называют чистой дисгенезией гонад или чистой дисгенезией яичников.
Это расстройство редко диагностируется у детей препубертатного возраста, т.к. наружные половые органы выглядят нормально, никаких др. аномалий не отмечается, рост в норме. В пубертатном возрасте половое созревание не происходит. Уровень гонадотропных гормонов в плазме повышен. Задержка закрытия эпифизарной зоны роста может привести к формированию евнухоидного телосложения. УЗИ ОМТ выявляет рудиментарные яичники.
Заболевание у сестер, кровное родство родителей и неспособность обнаружить мозаицизм указывают на АуР-тип наследования, ограниченный женским полом. Данное заболевание часто встречается в Финляндии (1:8300 живорожденных детей женского пола). В соответствии с проведенными наблюдениями, причинами заболевания в этой популяции считаются несколько мутаций в гене рецептора ФСГ (на хромосоме 2р). В противоположность этому, у мексиканских женщин с кариотипом 46,XX и дисгенезией гонад мутации гена рецептора ФСГ не выявлены. У некоторых пациентов ХХ-дисгенезия гонад сочеталась с нейросенсорной глухотой (синдром Перро). Также существует сообщение о пациентке с этим заболеванием и сопутствующим дефицитом гормона роста и вирилизацией. Существуют различные генетические формы этого расстройства.
Мюллерова агенезия или синдром Майера-Рокитанского-Кюстера-Хаузера — вторая наиболее распространенная причина первичной аменореи после дисгенезии гонад, встречающаяся у 1:4000-5000 женщин, описана в сочетании с дисгенезией гонад с кариотипом 46,XX у 17-летней девушки-подростка с первичной аменореей и отсутствием развития МЖ. Зарегистрирован один случай дисгерминомы с гигантскими синцитиотрофобластными клетками. У женщины 18 лет с первичной аменореей и отсутствием структур, развивающихся из мюллеровых протоков, односторонней агенезией почки и клиническими признаками гиперандрогении, с фенотипом, напоминающим синдром Майера-Рокитанского-Кюстера-Хаузера, обнаружена мутация с потерей функции в гене WNT4. Лечение состоит из заместительной терапии эстрогенами.
в) 45,Х/46,XY-дисгенезия гонад. 45,Х/46,XY-дисгенезия гонад, также называемая смешанной дисгенезией гонад, характеризуется крайне разнообразными постнатальными фенотипическими проявлениями в диапазоне от тернероподобного синдрома до мужского фенотипа с пенильной уретрой. Клинически можно выделить три основных фенотипа. Невысокий рост — основной симптом у всех детей, страдающих данным состоянием. В 90% пренатально диагностированных случаев отмечается нормальный мужской фенотип.
У некоторых пациентов отсутствуют признаки вирилизации, наблюдается женский фенотип, часто отмечаются соматические признаки синдрома Тернера. Это состояние диагностируется до наступления пубертатного периода, в случае проведения хромосомных исследований у низкорослых детей женского пола, или позже, когда хромосомные исследования проводятся по причине отсутствия полового созревания. Присутствуют маточные трубы и матка. Половые железы представлены недифференцированными тяжами, располагающимися в БП; хромосомное исследование тяжа часто выявляет клеточную линию с кариотипом ХУ. Половая железа, представленная в виде тяжа, несколько отличается от таковой у женщин с синдромом Тернера. Помимо волнистой соединительной ткани часто встречаются структуры в виде трубочек или жгутов, случайные скопления гранулезных клеток и клетки мезонефроса или хилюсные клетки.
У некоторых детей наблюдается умеренная вирилизация, проявляющаяся изолированной клиторомегалией в препубертатном возрасте. Структуры, в норме развивающиеся из мюллеровых протоков, присутствуют, но в период полового созревания происходит вирилизация. У этих пациентов часто обнаруживаются яичко, расположенное в БП, с противоположной стороны — половая железа, представленная в виде тяжа, и маточные трубы с двух сторон.
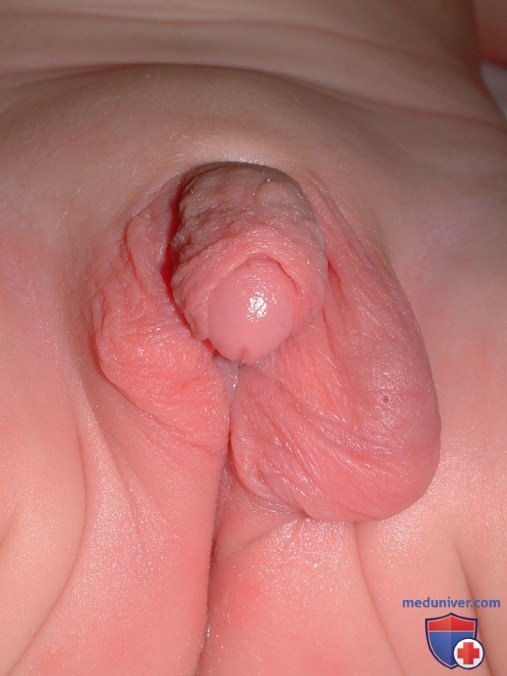
У многих детей с кариотипом 45,X/46,XY в младенчестве наблюдаются неоднозначно выглядящие половые органы (рис. 2). С одной стороны в губно-мошоночной складке обнаруживаются яичко и семявыносящий проток, а на противоположной стороне выявляется половая железа в виде тяжа. Несмотря на наличие яичка, с двух сторон часто присутствуют маточные трубы. Также часто обнаруживается инфантильная или рудиментарная матка.
Рисунок 2. У новорожденного с кариотипом 45,Х/46ХY, с расстройством полового развития, связанным с половой хромосомой, при рождении отмечено, что наружные половые органы соответствуют мужскому полу с пенисом размером 2,5 на 1,2 см и пеноскротальной гипоспадией. Слева в частично расщепленной мошонке пальпировалась половая железа, а справа половая железа не определялась. Биопсия половых желез выявила яичко с левой стороны и половую железу, представленную тяжом, с правой. Диагноз — смешанная дисгенезия гонад.
При смешанной дисгенезии гонад также описаны др. генотипы и фенотипы. У 25% из 200 обследованных пациентов присутствует дицентрическая У-хромосома (45,X/46,X,dic Y). У некоторых пациентов У-хромосома м.б. представлена только фрагментом (45,X/45,X+fra); применение У-специфических генетических зондов позволяет установить происхождение данного фрагмента. Неизвестно, почему один и тот же генотип (45,X/46,XY) может привести к развитию таких разнообразных фенотипов. У некоторых пациентов описаны мутации в гене SRY.
У детей с женским фенотипом не возникает проблемы с воспитанием. Пациентов с легкими проявлениями вирилизации до постановки диагноза воспитывают как девочек. Пациенты с неоднозначно выглядящими половыми органами часто клинически неотличимы от пациентов с различными типами нарушений формирования пола при кариотипе 46,XY. В некоторых случаях следует внимательно подойти к рассмотрению вопроса о воспитании. К факторам, которые могут повлиять на данное решение, относятся низкорослость, потребность в хирургической реконструкции половых органов, присутствие структур, развивающихся из мюллеровых протоков, а также необходимость хирургического удаления половых желез из-за предрасположенности к развитию в них ЗНО. У некоторых пациентов, наблюдаемых до достижения ими взрослого возраста, предположительно нормальное яичко оказывается дисгенетическим, с потерей в последующем функции клеток Лейдига и Сертоли.
При обследовании 22 пациентов со смешанной дисгенезией гонад не выявлено значимой связи или корреляции между внутренним и внешним фенотипами или эндокринной функцией и морфологическими характеристиками половых желез. При воспитании ребенка пол определялся по внешнему виду наружных половых органов. У 11 пациентов базальный уровень тестостерона и уровень тестостерона после стимуляции хорионическим гонадотропином человека были ниже, чем у субъектов в группе контроля.
Опухоли половых желез (гонадобластомы) встречаются у 25% этих детей. Локус гонадобластомы локализуется в области вблизи центромеры У-хромосомы (GBY). Развитию герминогенных опухолей предшествуют изменения, свойственные для карциномы in situ. У всех пациенток, воспитанных как женщины, следует удалить обе половые железы, а у пациентов, воспитанных как мужчины, — недифференцированную гонаду.
Не существует корреляции между долей линий клеток с кариотипом 45,X/46,XY, определяемой в крови и фибробластах, и фенотипом. В прошлом все пациенты привлекали внимание врачей по причине фенотипических аномалий. Однако при пренатальном обнаружении истинного хромосомного мозаицизма 45,X/46,XY-мозаицизм встречается у ~7% плодов. Из 76 младенцев с пренатально диагностированным 45,X/46,ХУ-мозаицизмом у 72 зарегистрирован нормальный мужской фенотип, у 1 — женский фенотип и у 3 детей мужского пола отмечалась гипоспадия. Из 12 детей мужского пола, чьи половые железы были исследованы, аномалии выявлены только в трех случаях. Эти данные необходимо учитывать при консультировании семьи с пренатально выявленным генотипом 45,X/46,XY у ребенка.
г) Женщины с кариотипом ХХХ, ХХХХ и ХХХХХ:
1. Женщины с кариотипом ХХХ. Хромосомная конституция 47,XXX (трисомия) считается наиболее частой хромосомной аномалией с наличием дополнительной Х-хромосомы у женщин. Встречается у 1:1000 живорожденных детей женского пола. В 68% случаев это состояние вызвано нерасхождением материнских хромосом в мейозе, но большинство случаев конституций 45,X и половина конституций 47,XXX вызваны ошибками в отцовской половой хромосоме. Фенотип — нормальный, женский; этих младенцев и детей невозможно распознать по внешнему виду гениталий.
Половое развитие и менархе в норме. В результате большинства беременностей рождаются нормальные дети. К 2 годам становятся очевидными задержки речевого и языкового развития, а в некоторых случаях наблюдается отсутствие координации, плохая успеваемость и инфантильное поведение. Такие женщины отличаются высоким ростом, у них наблюдаются расстройства поведения, и им часто требуется коррекционное образование. При МРТ высокого разрешения у 10 пациенток с кариотипом 47,XXX отмечались более низкие объемы миндалевидных тел, чем у 20 обследуемых с нормальным кариотипом в группе контроля. Также у 10 пациенток с кариотипом 47,XXY отмечены меньшие объемы миндалевидных тел. При обследовании 155 женщин 62% из них были физически нормальными. В пределах синдрома существует заметная вариабельность, и у небольшой доли женщин наблюдается хорошая координация, они социально адаптированы и достигают успехов в учебе.
2. Женщины с кариотипом ХХХХ и ХХХХХ. Большинство женщин с этими редкими кариотипами были умственно отсталыми. Сопутствующие пороки развития: эпикантальные складки, гипертелоризм, клинодактилия, поперечные ладонные складки, лучелоктевой синостоз и врожденные заболевания сердца. Половое созревание часто бывает неполным и может вообще не наступить. У трех женщин с синдромом тетрасомии Х-хромосомы наблюдались роды, но у женщин с кариотипом 49,ХХХХХ не было зарегистрировано ни одной беременности. Большинство женщин с кариотипом 48,ХХХХ высокого роста, при этом средний рост у них 169 см, а общая характерная фенотипическая черта при кариотипе 49,ХХХХХ — низкий рост.
д) Синдром Нунан. Женщины с синдромом Нунан демонстрируют определенные аномалии, также встречающиеся у женщин с синдромом Тернера (45,X), но у них нормальный хромосомный набор 46,XX. Наиболее распространенные аномалии — те же, что описаны у мужчин с синдромом Нунан. Невысокий рост — один из главных признаков данного синдрома. Фенотип отличается от такового при синдроме Тернера в нескольких отношениях. Часто присутствуют когнитивные нарушения, пороки сердца представлены стенозом легочного клапана или ДМПП, а не пороком развития аорты, нормальная половая зрелость с задержкой на 2 года, также поступали сообщения о развитии преждевременной недостаточности яичников. Лечение гормоном роста одобрено FDA для использования у пациентов с синдромом Нунан и низкорослостью.
е) Другие дефекты яичников. У некоторых молодых женщин без хромосомных аномалий половые железы представлены в виде тяжа, содержащего отдельные половые клетки или с полным отсутствием половых клеток. Уровень гонадотропных гормонов повышен. Цитотоксические ЛП, особенно алкилирующие ЛС (циклофосфамид и бусульфан, прокарбазин, этопозид), а также лучевая терапия на область яичников для лечения ЗНО считаются частыми причинами недостаточности яичников. На примере молодых женщин с лимфомой Ходжкина было продемонстрировано, что сочетание XT и лучевой терапии в области ОМТ может оказать больший вред, чем любой из этих методов лечения в отдельности. После облучения или комбинированной XT функция яичников чаще сохраняется или восстанавливается у подростков, чем у более взрослых женщин; после такого лечения возможно наступление нормальной беременности. Режимы лечения могут привести к определенной степени повреждения яичников у большинства женщин, получавших лечение по поводу ЗНО.
Средняя летальная доза для человеческой яйцеклетки - 4 Гр; дозы, равные 6 Гр, вызывают первичную аменорею. Транспозиция яичников перед облучением живота и таза в детском возрасте может сохранить функцию яичников при уменьшении дозы облучения до 4-7 Гр.
Недостаточность яичников аутоиммунного генеза встречается у 60% детей >13 лет с аутоиммунной полиэндокринопатией I типа (болезнь Аддисона, гипопаратиреоз, слизисто-кожный кандидоз). В мире это состояние, также известное как аутоиммунный полигландулярный синдром 1-го типа, встречается редко, кроме Финляндии, где в результате эффекта гена основателя оно встречается у 1:25 000 человек. Ген, ответственный за это расстройство, расположен на хромосоме 21 и связан с HLA DR5. У пациенток с аутоиммунным полигландулярным синдромом 1-го типа и недостаточностью яичников описана связь с HLA-A3. У девушек, страдающих данным заболеванием, может не наступать половое созревание, или у молодых женщин может возникать вторичная аменорея. В яичниках может наблюдаться лимфоцитарная инфильтрация, или яичники могут напоминать тяж. У большинства пациентов, страдающих этим заболеванием, обнаруживаются циркулирующие АТл к клеткам, участвующим в стероидогенезе, и аутоАТл к 21-гидроксилазе. У 5% из пациентов с аутоиммунными полигландулярными синдромами обнаружен гипогонадизм.
Такое состояние также возникает у молодых женщин, изолированно или в сочетании с др. аутоиммунными заболеваниями, что приводит к вторичной аменорее (преждевременной недостаточности яичников). Оно наблюдается у 0,2-0,9% женщин <40 лет. Преждевременная недостаточность яичников — гетерогенное заболевание, вызванное множеством факторов: хромосомных, генетических, ферментативных, инфекционных и ятрогенных. При сочетании с аутоиммунным заболеванием надпочечников присутствуют аутоАТл к клеткам, участвующим в стероидогенезе. Эти АТл связываются с ферментами P450scc, 17α-ОН или 21-ОН. При сочетании с рядом эндокринных и неэндокринных аутоиммунных заболеваний, а не с аутоиммунными заболеваниями надпочечников, аутоАТл к клеткам, участвующим в стероидогенезе, встречаются редко. У 10-39% взрослых пациенток с преждевременной недостаточностью яичников выявляется второе аутоиммунное заболевание, часто бессимптомное.
Такие заболевания могут включать аутоиммунное заболевание ЩЖ, СД-1, СКВ, ВЗК, ИТП или гемолитическую анемию, глютеновую энтеропатию, миастению и РА. У пациентки 17 лет с ИТП и кариотипом 47,XXX отмечалась преждевременная недостаточность яичников аутоиммунного генеза. У пациенток с преждевременной недостаточностью яичников нет нейрокогнитивных дефектов, в отличие от больных с синдромом Тернера.
Галактоземия, особенно классическая форма этого заболевания, приводит к повреждению яичников во в/утробном периоде. Уровни ФСГ и ЛГ повышены с раннего возраста. Повреждение яичников м.б. вызвано дефицитом уридиндифосфат-галактозы. Синдром Дениса-Драша, вызванный мутацией гена WT1, может привести к дисгенезии яичников.
Атаксия-телеангиэктазия может сочетаться с гипоплазией яичников и повышением уровня гонадотропных гормонов; причина этого неизвестна. У нескольких женщин развивались гонадобластомы и дисгерминомы.
Гипергонадотропный гипогонадизм возникает из-за резистентности яичников к эндогенным и к экзогенным гонадотропным гормонам (синдром Сэвиджа). Это состояние также встречается у женщин с преждевременной недостаточностью яичников. Это состояние м.б. вызвано присутствием антиовариальных АТл или аномалиями рецепторов к ФСГ. Известны сообщения об АуР-заболевании, характеризующемся мутацией гена рецептора к ФСГ. У нескольких женщин с хромосомным набором 46,XX, с клиническими проявлениями первичной аменореи и повышенным уровнем гонадотропных гормонов были обнаружены инактивирующие мутации гена рецептора к ЛГ. Это говорит о том, что для нормального развития фолликулов и овуляции необходимо воздействие ЛГ. Др. генетические нарушения, связанные с недостаточностью яичников, включают мутации в генах, кодирующих фактор транскрипции SF-1, FOXL2, GNAS, CYP17 и CYP19.