Лейкозы являются наиболее распространенными онкологическими заболеваниями у детей и подростков, на их долю приходится 31% всех ЗНО, возникающих у детей <15 лет. В США лейкоз диагностируется ежегодно у 3100 детей и подростков <20 лет, т.о., ежегодная заболеваемость составляет 4,5:100 000 детей.
На долю ОЛЛ приходится 77% случаев от общего количества детского лейкоза, острый миелобластный лейкоз (ОМЛ) составляет 11%, хронический миелолейкоз (ХМЛ) — 2-3% и ювенильный миеломоноцитарный лейкоз (ЮММЛ) — 1-2%. Остальные случаи представляют собой разл. формы острых и хронических лейкозов, которые не соответствуют классическим определениям ОЛЛ, ОМЛ, ХМЛ и ЮММЛ.
Лейкозы можно определить как группу ЗНО, при которых генетические аномалии в гемопоэтической клетке приводят к нерегулируемой клональной пролиферации клеток. Потомство этих клеток имеет преимущество в росте по сравнению с нормальными клеточными элементами из-за повышенной скорости пролиферации и сниженной скорости спонтанного апоптоза.
В результате возникает нарушение нормальной функции костного мозга и, в конечном итоге, развивается недостаточность костного мозга. Клинические картина, лабораторные данные и ответ на терапию зависят от вида лейкоза.
а) Острый лимфобластный лейкоз (ОЛЛ). Детский ОЛЛ стал первым излечимым диссеминированным ЗНО. ОЛЛ на самом деле представляет собой гетерогенную группу ЗНО с рядом характерных генетических аномалий, в результате которых возникают разл. клинические картины, при этом ответы на терапию также отличаются.
1. Эпидемиология. Ежегодно в США диагностируется — 3100 случаев ОЛЛ у детей и подростков <20 лет. Пик заболеваемости приходится на 2-3 года, при этом во всех возрастных группах ОЛЛ чаще наблюдается у мальчиков, чем у девочек. Среди белого населения социально-экономически развитых стран пик заболеваемости в этой возрастной группе был выявлен еще десятилетия назад, с тех пор эти данные подтвердились и для афроамериканского населения США.
Это заболевание чаще встречается у детей с определенными хромосомными аномалиями, такими как синдром Дауна (Down), синдром Блума (Bloom), атаксия-телеангиэктазия и анемия Фанкони (Fanconi). У однояйцевых близнецов риск для второго близнеца, если у одного из них развивается лейкоз, выше, чем в общей популяции. Если ОЛЛ диагностируется у первого близнеца в 1-й год жизни и близнецы имеют общую (монохориальную) плаценту, риск для второго близнеца составляет >70%.
Если ОЛЛ у первого близнеца развивается к 5-7 годам, то риск для второго близнеца как минимум вдвое выше, чем в общей популяции, независимо от зиготности.
2. Этиология. Практически во всех случаях этиология ОЛЛ неизвестна, хотя лейкоз детского возраста могут обуславливать несколько генетических и экологических факторов (табл. 1). Считается, что в большинстве случаев ОЛЛ развивается в результате соматических мутаций лимфоцитов, происходящих после оплодотворения. Однако обнаружение специфических для лейкоза последовательностей слияния генов в архивных образцах пятен крови, собранных для неонатального скрининга, у ряда детей с развитием ОЛЛ в более поздние сроки указывает на то, что в некоторых случаях для инициации злокачественного процесса значение имеют события, возникающие во время в/утробного развития.
Длительный латентный период до манифестации заболевания, наблюдаемый у некоторых детей, который, как сообщается, может продолжаться до 14 лет, подтверждает гипотезу о необходимости дополнительных к уже имеющимся генетическим модификаций для проявления заболевания. Более того, те же самые мутации были обнаружены в собранных для неонатального скрининга пятнах крови детей, у которых лейкоз так никогда и не развился.
Воздействие диагностического излучения как в/утробно, так и в детском возрасте повышает показатели заболеваемости ОЛЛ. Кроме того, опубликованные отчеты и исследования географических кластеров заболевания вызывают обеспокоенность, т.к. факторы окружающей среды могут способствовать повышению показателей заболеваемости ОЛЛ. На сегодняшний день в США не было выявлено никаких др. факторов окружающей среды, кроме радиации. В некоторых развивающихся странах существует связь между В-клеточным ОЛЛ и инфекционными заболеваниями, вызванными ВЭБ.
3. Цитологическая классификация. Классификация ОЛЛ основывается на выявлении характерных особенностей злокачественных клеток в костном мозге для определения их морфологии и фенотипа, который измеряется мембранными маркерами клеток, а также для определения их цитогенетических и молекулярно-генетических особенностей. Для постановки диагноза обычно бывает достаточно только морфологического исследования, но для более точной классификации заболевания необходимы и др. исследования, т.к. это может оказать существенное влияние на прогноз и выбор соответствующей терапии. В настоящее время используется классификация лейкозов ВОЗ.
Поверхностные маркеры показывают, что 85% всех случаев классифицируются по фенотипу как В-клеточный лимфобластный лейкоз (ранее называемый ОЛЛ из предшественников В-клеток/пре-В-ОЛЛ), 15% случаев представляют собой Т-клеточный лимфобластный лейкоз, и 1% происходит из зрелых В-клеток. Редко встречающийся лейкоз из зрелых В-клеток называется лейкозом Беркитта (Burkitt); он представляет собой один из видов наиболее быстро развивающихся ЗНО человека и требует иного терапевтического подхода, чем др. подтипы ОЛЛ.
У небольшого процента детей с лейкозом наблюдаются поверхностные маркеры, характерные как для лимфоидной, так и миелоидной линии кроветворения.
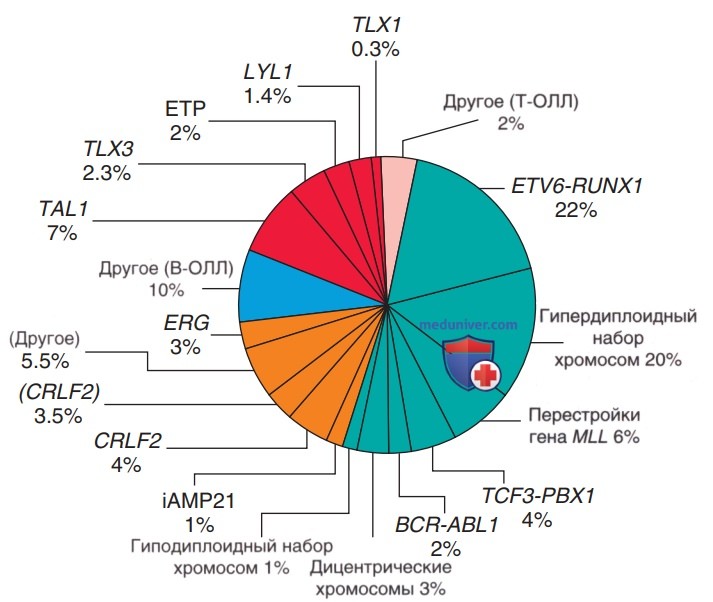
В соответствии с хромосомными аномалиями ОЛЛ разделяется на прогностические подгруппы (табл. 2). На сегодняшний день уже известны многие генетические альтерации, включая инактивацию генов-супрессоров опухолей и мутации, активирующие сигнальные пути NOTCH1/RAS; в будущем эти данные м.б. использованы в клинической практике (рис. 1).
Рисунок 1. Расчетная частота встречаемости специфических генотипов при остром лимфобластном лейкозе (ОЛЛ) детского возраста. Синие секторы диаграммы относятся к В-клеточному ОЛЛ (В-ОЛЛ), желтые секторы обозначают недавно идентифицированные подтипы В-клеточного ОЛЛ, а красные секторы — Т-клеточный ОЛЛ (Т-ОЛЛ)
Молекулярно-генетические аномалии м.б. обнаружены с помощью таких методов, как ПЦР и флуоресцентная гибридизация in situ, также эти методы, чья клиническая эффективность доказана, могут применяться для обнаружения даже небольшого количества злокачественных клеток как при диагностике, так и во время последующего наблюдения [минимальная остаточная болезнь (МОБ), см. далее].
Технология ДНК-микрочипов позволяет анализировать экспрессию тысяч генов в лейкозной клетке. Эта технология открывает возможности для более глубокого изучения фундаментальной биологии и помогает выработать терапевтическую стратегию для лечения ОЛЛ.
4. Клинические проявления. Начальные проявления ОЛЛ обычно неспецифичны и носят относительно кратковременный характер. Часто наблюдаются анорексия, повышенная утомляемость, общее недомогание, нервозность и перемежающаяся субфебрильная ТТ. Могут присутствовать боли в костях/суставах, особенно в нижних конечностях.
В более редких случаях симптомы могут наблюдаться в течение нескольких месяцев, могут локализоваться преимущественно в костях/суставах, может наблюдаться отек сустава. Боль в костях сильная и может будить пациента по ночам.
По мере прогрессирования заболевания более очевидными становятся признаки и симптомы недостаточности костного мозга, т.е. бледность, повышенная утомляемость, непереносимость ФН, кровоподтеки, кровотечения из слизистой оболочки полости рта/носа, а также лихорадка, которая м.б. вызвана инфекцией/самим заболеванием.
Инфильтрация органов может вызвать лимфаденопатию, гепатоспленомегалию, увеличение яичек и поражение ЦНС (поражение ЧМН, головная боль, судороги). РДС может развиваться в результате тяжелой анемии/сдавления ДП ЛУ средостения.
Наблюдаемые при физикальном осмотре бледность, вялость, пурпурные и петехиальные поражения кожи/кровоизлияния в слизистые оболочки могут указывать на недостаточность костного мозга. Пролиферативный характер заболевания может проявляться в виде лимфаденопатии, спленомегалии и, реже, гепатомегалии. Пациенты с поражением костей/суставов могут жаловаться на острые болезненные ощущения при дотрагивании до кожи, расположенной над костью, а при объективном исследовании может наблюдаться припухлость суставов/выпот в полости суставов.
Однако при поражении костного мозга боль в костях м.б. глубокой, а болезненность при дотрагивании может отсутствовать. В редких случаях у пациентов наблюдаются признаки повышенного ВЧД, свидетельствующие о лейкозном поражении ЦНС. К таким признакам относятся отек диска зрительного нерва (см. рис. 520.3), кровоизлияния в сетчатку глаза и параличи ЧМН. РДС обычно обусловлен анемией, но может развиваться из-за наличия большой опухолевой массы в переднем средостении (напр., при поражении тимуса/ЛУ).
Такое поражение чаще всего наблюдается у мальчиков подросткового возраста с Т-клеточным ОЛЛ. При Т-клеточном ОЛЛ количество лейкоцитов обычно более высокое.
В-лимфобластный лейкоз является наиболее распространенным иммунофенотипом, манифестация этого заболевания происходит в 1-10 лет. Медиана числа лейкоцитов при первичном обследовании составляет 33 000/мкл, хотя у 75% пациентов количество лейкоцитов <20 000/мкл; тромбоцитопения наблюдается у 75% пациентов, а гепатоспленомегалия — у 30-40%.
При всех лейкозах симптомы поражения ЦНС наблюдаются при первичном обследовании у 5% пациентов (у 10-15% в СМЖ обнаруживаются бластные клетки). При постановке диагноза редко наблюдаются симптомы поражения яичек, но проведенные ранее исследования свидетельствуют о вовлечении яичек у 25% мальчиков. Показаний к проведению биопсии яичек нет.
5. Диагностика. Диагноз ОЛЛ убедительно подтверждается показателями периферической крови, указывающими на недостаточность костного мозга. У большинства пациентов наблюдается анемия и тромбоцитопения. Лейкозные клетки м.б. не обнаружены в мазках периферической крови при рутинных лабораторных исследованиях.
Общее количество лейкоцитов у многих пациентов с ОЛЛ составляет <10 000/мкл. В таких случаях лейкозные клетки часто первоначально считаются «атипичными лимфоцитами», и только при дальнейших исследованиях выясняется, что эти клетки оказываются частью опухолевого клона.
Когда результаты анализа периферической крови указывают на возможность лейкоза, следует незамедлительно провести исследования костного мозга для постановки точного диагноза. Важно, чтобы были проведены все исследования, необходимые для подтверждения диагноза и адекватной классификации типа лейкоза, включая аспирацию и биопсию костного мозга, проточную цитометрию, цитогенетику и молекулярные исследования.
Диагноз ОЛЛ ставится на основании исследования костного мозга, которое показывает, что >25% клеток костного мозга представляют собой однородную популяцию лимфобластов. Первичное обследование также включает исследование СМЖ. Если в СМЖ обнаруживаются лимфобласты и количество лейкоцитов повышено, это свидетельствует о лейкозе с поражением ЦНС/менингеальных оболочек. Такие результаты исследования указывают на более позднюю стадию заболевания и на необходимость дополнительной терапии ЦНС и системной терапии.
Люмбальная пункция для стадирования заболевания м.б. выполнена вместе с введением первой дозы интратекальной XT, если диагноз лейкоза был установлен ранее на основании исследования костного мозга. Первичная люмбальная пункция должна проводиться опытным специалистом, поскольку травматическая процедура повышает риск рецидива с поражением ЦНС.
6. Дифференциальная диагностика. Диагноз лейкоза быстро подтверждается у пациентов с типичными признаками и симптомами, такими как анемия, тромбоцитопения, повышенное количество лейкоцитов и наличие бластных клеток в мазке периферической крови. Повышение уровня ЛДГ часто указывает на ОЛЛ. При наличии только панцитопении следует исключить апластическую анемию (врожденную/приобретенную), миелофиброз и семейный гемофагоцитарный лимфогистиоцитоз.
Патология одной клеточной линии, наблюдаемая при транзиторной эритробластопении детского возраста, иммунной тромбоцитопении и врожденной/приобретенной нейтропении, редко является проявлением ОЛЛ. Для ДД ОЛЛ от инфекционного мононуклеоза у пациентов с острой лихорадкой и лимфаденопатией и от ЮИА у пациентов с лихорадкой, отеком суставов, болью в костях, но часто без болезненности при пальпации, требуется большая настороженность. При наличии таких симптомов может потребоваться исследование костного мозга.
ОЛЛ должен быть дифференцирован от ОМЛ и др. ЗНО, таких как нейробластома, рабдомиосаркома, саркома Юинга (Ewing) и ретинобластома, которые поражают костный мозг и могут иметь клинические проявления и лабораторные показатели, сходные с показателями при ОЛЛ.
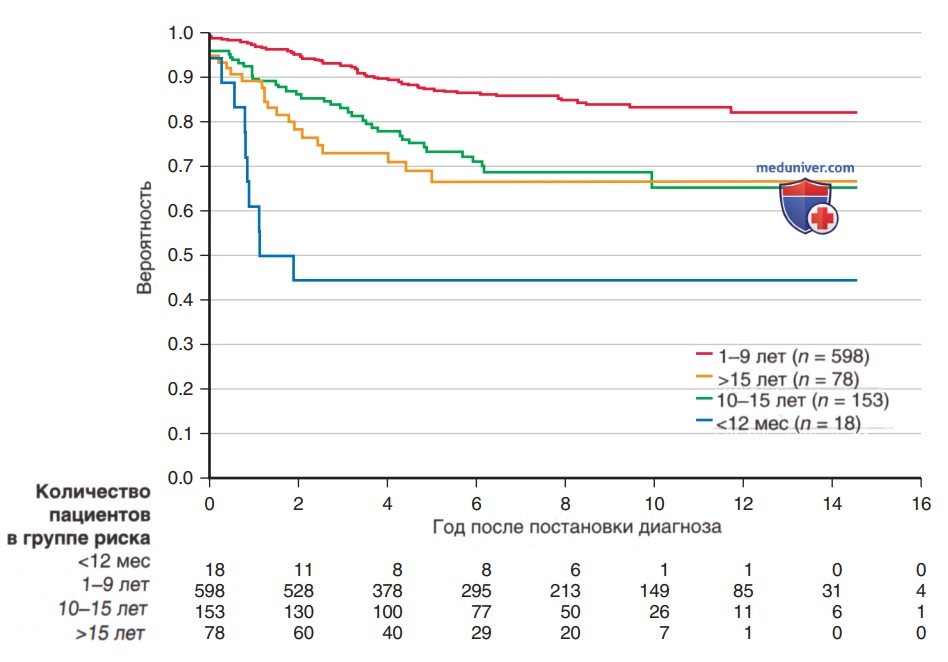
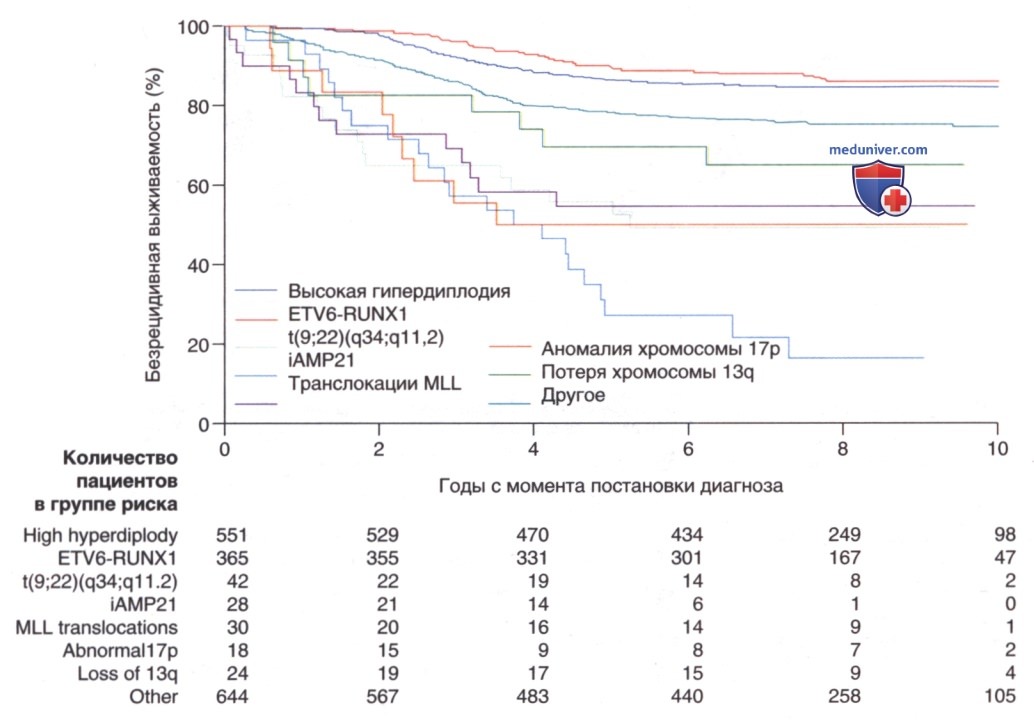
7. Лечение. Лечение ОЛЛ является наиболее важным прогностическим фактором: без эффективной терапии эта болезнь смертельна. Показатели общей выживаемости у детей с ОЛЛ стали значительно улучшаться с 1970-х годов благодаря использованию мультиагентных XT-схем, интенсификации терапии и подбору метода лечения с учетом риска рецидива (рис. 2). Выживаемость также обусловлена возрастом (рис. 3) и подтипом заболевания (рис. 4).
Рисунок 2. Знаковые достижения в области лечения острого лимфобластного лейкоза (ОЛЛ) детского возраста. Показатели общей пятилетней выживаемости педиатрических пациентов с ОЛЛ, представленные на основе данных программы SEER 1 — «Течение, распространенность и исходы злокачественных новобразований», изображены на графике в соотношении со знаковыми достижениями в области лечения (обозначены белыми кружками; левая таблица) и в понимании биологии ОЛЛ детского возраста (обозначены черными кружками; правая таблица)
Рисунок 3. Анализ бессобытийной выживаемости по методу Каплана-Мейера в зависимости от возраста пациента на момент постановки диагноза острого лимфобластного лейкоза
Рисунок 4. Анализ безрецидивной выживаемости по методу Каплана-Мейера в зависимости от биологического подтипа лейкоза
Риск-ориентированная терапия является современным стандартом лечения ОЛЛ и учитывает возраст пациента на момент постановки диагноза, исходное количество лейкоцитов, иммунофенотипические и цитогенетические характеристики популяций бластов, скорость раннего ответа на лечение (т.е. насколько быстро костный мозг/периферическая кровь м.б. санированы от лейкозных клеток) и оценку МОБ в конце индукционной терапии. Для определения степени риска развития ОЛЛ разл. исследовательские группы используют разл. факторы.
Национальный институт онкологии относит детей 1-10 лет и с уровнем лейкоцитов <50 000/мкл к группе стандартного риска. Дети <1 года и >10 лет/с исходным уровнем лейкоцитов >50 000/мкл относятся к группе высокого риска. Др. особенности, которые «-» влияют на исход заболевания, включают Т-клеточный иммунофенотип/медленный ответ на стартовую терапию. Хромосомные аномалии, включая гиподиплоидию, филадельфийскую хромосому и перестройки гена КМТ2А (MLL), предвещают неблагоприятный исход.
Др. мутации, напр., мутации в гене 1KZF1, как было установлено, сопряжены с неблагоприятным прогнозом и могут иметь значение для разработки алгоритмов лечения в будущем. Более благоприятные характерные особенности включают быстрый ответ на терапию, гипердиплоидию, трисомию специфических хромосом (4, 10 и 17) и перестройку генов ETV6-RUNX1 (ранее TEL-AMU).
Исход для пациентов из группы высокого риска м.б. улучшен за счет назначения более интенсивной XT, несмотря на большую ее токсичность. Риск рецидива у младенцев с ОЛЛ, как и у пациентов, имеющих специфические хромосомные аномалии, такие как t(4;11), еще более высокий, несмотря на интенсивную XT. Однако неблагоприятный исход при ОЛЛ с филадельфийской хромосомой t(9;22) был кардинально изменен благодаря добавлению иматиниба к интенсивной XT.
Иматиниб — это ЛП, специально разработанный для ингибирования BCR-ABL-киназы, возникающей в результате хромосомной транслокации. При таком подходе показатели бессобытийной выживаемости увеличились с 30 до 70%. Клинические испытания показывают, что прогноз для пациентов с медленным ответом на стартовую терапию м.б. улучшен назначением более интенсивной XT, нежели та, которая требуется пациентам с быстрым ответом.
Большинство детей с ОЛЛ проходят лечение в условиях клинических исследований, проводимых национальными/международными кооперативными группами. Стандартное лечение включает XT 2-3 года, при этом большинство пациентов достигает ремиссии в конце индукционной фазы. Пациенты в стадии клинической ремиссии могут иметь МОБ, которая м.б. обнаружена только с помощью специфических молекулярных ДНК-зондов для детектирования транслокаций и др.
ДНК-маркеров, содержащихся в лейкозных клетках/специализированного метода проточной цитометрии. МОБ может оцениваться количественно, а ее показатели могут использоваться для определения количества лейкозных клеток, присутствующих в костном мозге. Более высокие уровни МОБ, наблюдаемые в конце индукционной фазы, свидетельствуют о плохом прогнозе и более высоком риске последующего рецидива. Уровень МОБ >0,01% в костном мозге на 29-й день индукционной фазы является значимым фактором риска более короткой бессобытийной выживаемости для всех групп риска по сравнению с пациентами с «-» значениями МОБ.
Наличие признаков МОБ в конце индукционной фазы позволяет применять более интенсивное лечение ОЛЛ.
Стартовая терапия, называемая индукцией ремиссии, направлена на уничтожение лейкозных клеток в костном мозге. Во время этой фазы терапия длится 4 нед и состоит из еженедельного введения винкристина, ГКС, напр., дексаметазон/преднизон, и обычно однократной дозы пегилированной аспарагиназы длительного действия. Пациенты с высоким риском также получают даунорубицин с недельными интервалами. При таком подходе у 98% пациентов наступает стадия ремиссии, которая определяется по показателям уровня бластов в костном мозге (<5%) и возвращением количества нейтрофилов и тромбоцитов почти на нормальный уровень после 4-5 нед лечения. Интратекальная XT всегда проводится в начале лечения и по крайней мере еще один раз во время индукционной фазы.
Вторая фаза лечения, фаза консолидации, направлена на интенсивную терапию ЦНС в комбинации с продолжающейся интенсивной системной терапией с целью предотвращения последующих рецидивов со стороны ЦНС. Интратекальная XT проводится методом повторных люмбальных пункций. Т.о., вероятность более позднего рецидива со стороны ЦНС снижается до <5%, по сравнению с прошлыми уровнями частоты в 60%. Небольшому проценту пациентов с признаками, указывающими на высокий риск рецидива со стороны ЦНС, может проводиться облучение ГМ во время более поздних фаз лечения.
К этой категории относятся пациенты, на момент постановки диагноза имеющие лимфобласты в СМЖ и повышенное количество лейкоцитов в СМЖ/физикальные признаки нейролейкемии, такие как паралич ЧМН.
Соответственно, многие схемы лечения предписывают проведение терапии 14-28 нед, причем используемые ЛП и схемы варьируют в зависимости от группы риска пациента. Эта фаза лечения часто называется фазой интенсификации и включает фазы агрессивного лечения (отсроченная интенсификация), а также фазы лечения относительно нетоксичными ЛП (фаза промежуточной поддержки).
Мультиагентная XT, включающая такие ЛП, как цитарабин, метотрексат, аспарагиназа и винкристин, применяется во время этих фаз для ликвидации остаточной болезни.
Наконец, наступает фаза поддерживающего лечения, которая длится 2-3 года, в зависимости от используемого протокола. Пациентам назначают меркаптопурин ежедневно и метотрексат внутрь еженедельно, как правило, с интервальным введением винкристина и ГКС.
Во время первой ремиссии небольшому количеству пациентов с особенно плохим прогнозом, таким, как пациенты с неэффективной индукцией/экстремальной гиподиплоидией, м.б. показана трансплантация костного мозга в первой ремиссии.
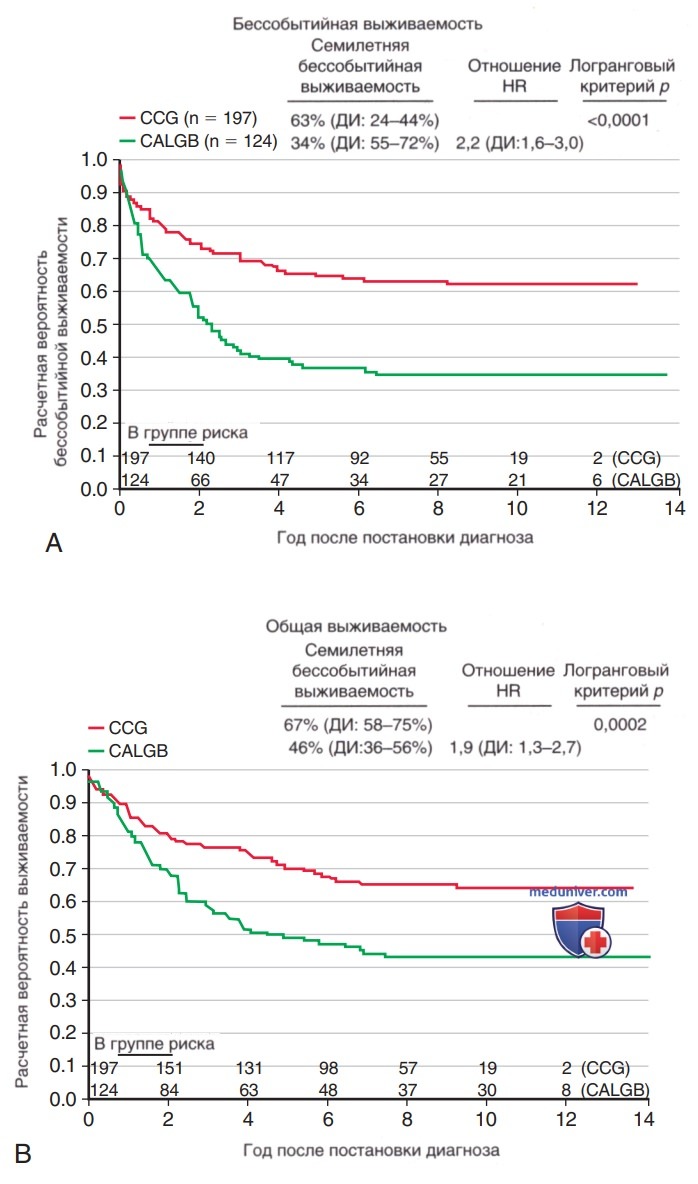
Подростки и молодые люди с ОЛЛ имеют худший прогноз по сравнению с детьми <15 лет. Они часто имеют неблагоприятные прогностические факторы и нуждаются в более интенсивной XT. Хороший исход пациенты этой возрастной группы имеют при лечении по педиатрическим, а не взрослым протоколам (рис. 5). Хотя данное наблюдение может объясняться многими факторами, остается важной терапия по педиатрическим протоколам, лучше всего в детском онкологическом центре.
Рисунок 5. Сравнение бессобытийной выживаемости у пациентов Группы В по исследованию рака и острого лейкоза (CALGB) (протокол лечения у взрослых, зеленая линия) и Детской онкологической группы (CCG) (протокол лечения у детей, красная линия)
Генетические полиморфизмы ферментов, играющих важную роль в метаболизме ЛП, могут оказывать влияние как на эффективность, так и на токсичность ХТ-ЛП. Фармакогенетическое тестирование гена тиопурин-S-метилтрансферазы, который кодирует один из ферментов метаболизма меркаптопурина, может выявить пациентов с диким типом полиморфизма (нормальная ферментная активность), с гетерозиготным полиморфизмом (ферментная активность незначительно снижена) и гомозиготным полиморфизмом (ферментная активность низкая/отсутствует).
Снижение ферментной активности тиопурин-6-метилтрансферазы приводит к накоплению токсического метаболита меркаптопурина и тяжелой миелосупрессии, требующей снижения дозы XT. В будущем лечение также м.б. стратифицировано по профилям экспрессии генов лейкозных клеток. В частности, на основании массивов экспрессии генов, индуцированных воздействием XT-агента, можно спрогнозировать, у каких пациентов ОЛЛ окажется резистентным.
- Лечение рецидивов. Основным препятствием для достижения «+» исхода заболевания является его рецидив. Результаты лечения пациентов с рецидивами остаются неутешительными, причем наиболее важными прогностическими показателями являются время после постановки диагноза и место рецидива заболевания. Кроме того, прогностическое значение имеют и др. факторы, такие как иммунофенотип (прогноз при Т-клеточном ОЛЛ хуже, чем при В-клеточном ОЛЛ) и возраст пациента при постановке первичного диагноза.
Рецидив в костном мозге возникает у 15-20% пациентов с ОЛЛ, что влечет за собой серьезнейшие последствия, особенно если рецидив возникает во время/вскоре после завершения терапии. Интенсивная XT ЛП, ранее не применявшимися у пациента, с последующей аллогенной трансплантацией стволовых клеток может привести к длительной выживаемости некоторых пациентов с костно-мозговым рецидивом. Все большую роль в лечении пациентов с рецидивом ОЛЛ будет играть терапия Т-клетками с химерным АГн-рецептором.
Частота рецидивов со стороны ЦНС снизилась до <5% с момента введения профилактической терапии ЦНС. Рецидив в ЦНС м.б. обнаружен во время проведения рутинной люмбальной пункции у бессимптомного пациента. У симптоматичных пациентов с рецидивом со стороны ЦНС обычно наблюдаются признаки повышенного ВЧД, м.б. изолированные параличи ЧМН. Диагноз подтверждается при обнаружении лейкозных клеток в СМЖ. Лечение включает интратекальную медикаментозную терапию и краниальное/краниоспинальное облучение. Также должна быть назначена системная XT, т.к. такие пациенты входят в группу высокого риска по развитию костномозгового рецидива. Большинство пациентов с рецидивом лейкоза, ограниченным поражением ЦНС, хорошо лечатся, особенно это касается пациентов с рецидивом в ЦНС через >18 мес после начала XT.
Тестикулярный рецидив встречается у <2% мальчиков с ОЛЛ, как правило, после завершения терапии. Такой рецидив возникает в виде безболезненного отека одного/обоих яичек. Диагноз подтверждается результатами биопсии пораженного яичка. Лечение включает системную XT и по возможности местное облучение. У высокого процента мальчиков с тестикулярным рецидивом повторное лечение м.б. успешным, выживаемость этих пациентов хорошая.
Самую свежую информацию о лечении ОЛЛ детского возраста можно получить из базы данных для врачебных запросов на веб-сайте Национального института онкологии.
8. Сопроводительная терапия. Успешное проведение агрессивных XT-программ в большой степени зависит от сопроводительной терапии. Пациенты с высоким уровнем лейкоцитов особенно подвержены риску развития синдрома лизиса опухоли в начале терапии. Профилактику/лечение почечной недостаточности, сопряженной с очень высоким уровнем мочевой кислоты в сыворотке крови, можно проводить аллопуринолом/уратоксидазой*. XT часто вызывает тяжелую миелосупрессию, которая может потребовать проведения гемотрансфузии эритроцитов и тромбоцитов, всегда требует высокой настороженности и агрессивной эмпирической антимикробной терапии сепсиса у детей с лихорадкой и нейтропенией. Во время проведения XT и в течение нескольких месяцев после ее завершения пациенты должны получать профилактическое лечение пневмоцистной пневмонии, вызванной Pneumocystis jiroveci.
P.S. * ЛП не зарегистрирован в РФ.
Успешное лечение ОЛЛ является прямым результатом интенсивной терапии, которая часто проводится токсичными ЛП. Однако такая интенсивная терапия может привести к большим проблемам в сфере образования, физического и психосоциального развития детей с ОЛЛ, повлечь за собой значительные финансовые затраты и стресс для их семей. Могут возникать как отдаленные, так и острые токсические эффекты. Для минимизации осложнений и достижения оптимального результата лечения требуется сотрудничество множества специалистов по лечению онкологических заболеваний, имеющих специальную подготовку и опыт в решении бесчисленного количества проблем, которые могут возникнуть во время ведения заболевания.
9. Прогноз. Усовершенствование методик лечения и стратификации рисков привело к значительному повышению выживаемости, причем текущие данные показывают, что общая пятилетняя выживаемость составляет 90% (рис. 6). Тем не менее, у пациентов, выживших после онкологического заболевания, с большей вероятностью развивается серьезная хроническая патология по сравнению с их братьями и сестрами, такие состояния могут включать заболевания скелетно-мышечной системы, болезни сердца и неврологические заболевания. В целом долгосрочное наблюдение за пациентами с ОЛЛ должно проводиться в клинике, где сопровождение детей и подростков может осуществляться разными специалистами для полного охвата проблем этих особых пациентов.
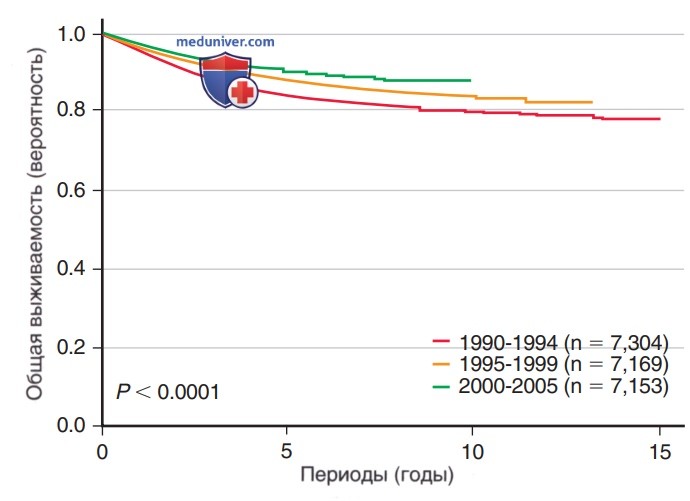
Рисунок 6. Оценка вероятности общей выживаемости пациентов с острым лимфобластным лейкозом по результатам исследования Детской группы онкологии, рассчитанная по периодам лечения 1990-1994, 1995-1999 и 2000-2005 гг.
б) Острый миелобластный лейкоз:
1. Эпидемиология. ОМЛ составляет 11% всех случаев детского лейкоза в США; ежегодно ОМЛ диагностируется у 370 детей. Относительная частота встречаемости ОМЛ увеличивается в подростковом возрасте, составляя 36% случаев лейкоза в возрастной группе 15-19 лет. Острый промиелоцитарный лейкоз — подтип ОМЛ, чаще встречающийся в некоторых регионах, в то время как заболеваемость др. подтипами в целом одинакова по всему миру. Было выявлено несколько хромосомных аномалий при ОМЛ, но предрасполагающих генетических/экологических факторов у большинства пациентов выявлено не было (см. табл. 1).
Тем не менее, был определен ряд факторов риска, которые включают ионизирующее излучение, ХТ-ЛП (напр., алкилирующие агенты, эпиподофиллотоксин), органические р-рители, пароксизмальную ночную гемоглобинурию и некоторые синдромы, такие как синдром Дауна, анемия Фанкони, синдром Блума, синдром Костмана, синдром Швахмана-Даймонда, синдром Даймонда-Блэкфана, синдром Ли-Фраумени и нейрофиброматоз 1-го типа.
2. Цитологическая классификация. Характерной особенностью ОМЛ является то, что при проведении аспирации/биопсии >20% клеток костного мозга в мазке-отпечатке представляют собой достаточно однородную популяцию бластных клеток с признаками, сходными с клетками крови миелоидного, моноцитарного и мегакариоцитарного рядов на ранних этапах дифференцировки. Современная практика требует применения проточной цитометрии для идентификации АГн клеточной поверхности и использования хромосомных и молекулярно-генетических методов для более точной диагностики и оптимального выбора метода лечения.
ВОЗ предложила новую систему классификации, основанную на морфологических особенностях, типах хромосомных аномалий и специфических генных мутаций. Такая система классификации содержит важную информацию о биологии и прогнозе ОМЛ (табл. 3).
3. Клинические проявления. Проявление симптомов и признаков ОМЛ является результатом замещения костного мозга злокачественными клетками и вызвано вторичной костномозговой недостаточностью. У пациентов с ОМЛ может наблюдаться любой из симптомов, обусловленных костномозговой недостаточностью при ОЛЛ/все эти симптомы. Кроме того, у пациентов с ОМЛ присутствуют признаки и симптомы, которые редко встречаются при ОЛЛ, включая п/к узелки/поражения по типу «черничного маффина» (особенно у младенцев), инфильтрацию десны (особенно при моноцитарных подтипах), признаки и лабораторные данные ДВС-синдрома (чаще всего указывают на острый промиелоцитарный лейкоз) и отдельные опухолевые массы, известные как хлоромы/гранулоцитарные саркомы.
Эти массы могут возникать при отсутствии выраженного поражения костного мозга и, как правило, обусловлены транслокацией t(8;21). Хлоромы могут развиваться в области орбиты глаза и в эпидуральном пространстве.
4. Диагностика. Анализ аспиратов и биоптатов костного мозга пациентов с ОМЛ обычно выявляет признаки гиперцеллюлярного костного мозга с однообразным клеточным составом. Метод проточной цитометрии и использование специальных красителей помогают идентифицировать клетки, содержащие миелопероксидазу, тем самым подтверждая диагноз лейкоза и его миелогенное происхождение. Некоторые хромосомные аномалии и молекулярно-генетические маркеры характерны для специфических подтипов заболевания (табл. 4).
5. Прогноз и лечение. Агрессивная мультиагентная ХТ успешно индуцирует ремиссию у 85-90% пациентов. Уровень выживаемости значительно вырос с 1970-х годов, когда выживало только 15% вновь диагностированных пациентов, по сравнению с уровнем выживаемости 60-70% при использовании современных методов лечения (рис. 7). Существуют разл. схемы индукционной ХТ, обычно включающие [антрациклин + высокие дозы цитарабина]. Эффективной может оказаться таргетная терапия, направленная на генетические маркеры (см. табл. 4). До 5% пациентов умирают от инфекции/кровотечения до достижения ремиссии. Постремиссионная терапия выбирается на основе комбинации цитогенетических и молекулярных маркеров лейкоза, а также ответа на индукционную ХТ (оценка МОБ).
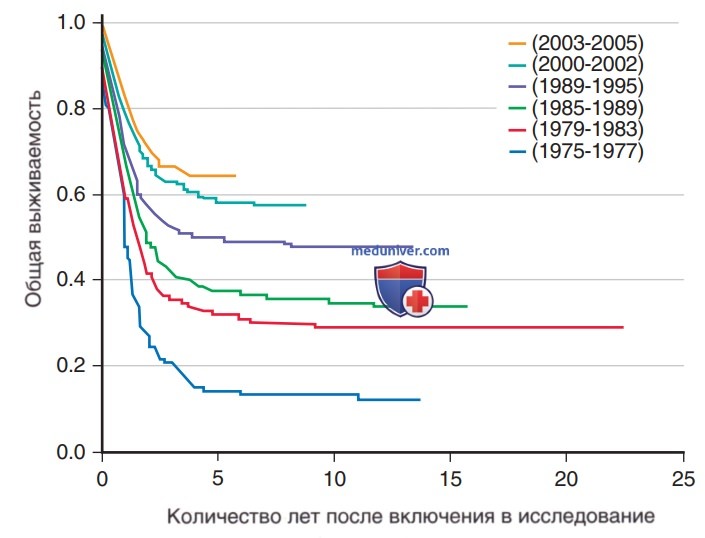
Рисунок 7. Показатели общей выживаемости, демонстрирующие постепенное за последние 40 лет улучшение результатов лечения острого миелобластного лейкоза детского возраста по данным исследований Группы детской онкологии и предыдущих долгосрочных исследований
Некоторым пациентам с благоприятными прогностическими характеристиками [t(8;21); t(15;17); inv(16)] и хорошим исходом при проведении только ХТ трансплантация стволовых клеток рекомендуется только после рецидива. Однако для пациентов с неблагоприятными прогностическими признаками (напр., с моносомией 7 и 5, аномалиями 5q- и 11q23) и плохими результатами ХТ трансплантация стволовых клеток может оказаться эффективной при первой ремиссии. С развитием методов сопроводительной терапии больше нет существенной разницы между показателями смертности при сравнении родственных и неродственных трансплантаций HLA-совместимых стволовых клеток при ОМЛ.
Острый промиелоцитарный лейкоз, характеризующийся перестройкой генов с участием рецептора ретиноевой кислоты [t(15;17); PML-RARA], очень чувствителен к полностью-трансретиноевой кислоте (третиноин) в сочетании с антрациклинами и цитарабином. При «+» результате этой терапии трансплантация костного мозга в первой ремиссии пациентам с острым промиелоцитарным лейкозом не нужна. Мышьяка триоксид — эффективный нецитотоксический ЛП для острого промиелоцитарного лейкоза. Данные исследований у взрослых показывают многообещающие результаты с использованием комбинированной терапии [третиноин + мышьяка триоксид] без цитотоксических ЛП в качестве стартовой терапии данного вида лейкоза и поддерживают идею нового исследования такого терапевтического режима у детей.
Пациентам с ОМЛ необходима усиленная сопроводительная терапия, поскольку интенсивное лечение, которое они получают, приводит к длительному подавлению функции костного мозга с очень высоким уровнем заболеваемости тяжелыми инфекциями, особенно стрептококковым сепсисом, вызванным Streptococcus viridans, и грибковыми инфекциями. Таким пациентам могут потребоваться длительная госпитализация, лечение филграстимом (гранулоцитарным колониестимулирующим фактором) и профилактические антимикробные ЛП.
Самую свежую информацию о лечении ОМЛ можно получить из базы данных для врачебных запросов на веб-сайте Национального института онкологии.
в) Синдром Дауна, острый лейкоз и транзиторное миелопролиферативное заболевание. Острый лейкоз встречается в 15-20 раз чаще у детей с синдромом Дауна, чем в общей популяции. Соотношение ОЛЛ и ОМЛ у пациентов с синдромом Дауна такое же, как и в общей популяции. Исключение составляет период первых 3 лет жизни, в это время ОМЛ встречается чаще. У детей с синдромом Дауна и ОЛЛ ожидаемый результат лечения немного менее благоприятный, чем у др. детей. Такое различие м.б. частично объяснено отсутствием благоприятных прогностических характеристик, таких как ETV6-RUNX1 и трисомий, а также генетическими аномалиями, обуславливающими неблагоприятный прогноз, такими как IKZF1.
Пациенты с синдромом Дауна демонстрируют выраженную чувствительность к метотрексату и др. антиметаболитам, что приводит к высокой токсичности при введении стандартных доз. Однако пациенты с синдромом Дауна и ОМЛ имеют гораздо более благоприятный исход, чем дети без синдрома Дауна, с уровнем длительной выживаемости >80%. После индукционной терапии эти пациенты получают менее интенсивную терапию, чтобы уменьшить токсичность, сохраняя при этом высокую эффективность лечения.
У 10% новорожденных с синдромом Дауна развивается транзиторный лейкоз/миелопролиферативное заболевание, характеризующееся высоким содержанием лейкоцитов, бластных клеток в периферической крови и связанной с ними анемией, тромбоцитопенией и гепатоспленомегалией. Эти симптомы обычно разрешаются в первые 3 мес жизни. Хотя такие новорожденные могут нуждаться во временной трансфузионной поддержке, им не требуется XT при отсутствии явных признаков осложнений, опасных для жизни. Однако пациенты с синдромом Дауна и транзиторным миелопролиферативным заболеванием нуждаются в тщательном наблюдении, поскольку у 20-30% из них к 3 годам (средний возраст начала заболевания — 16 мес) развивается типичный лейкоз (часто острый мегакариоцитарный лейкоз).
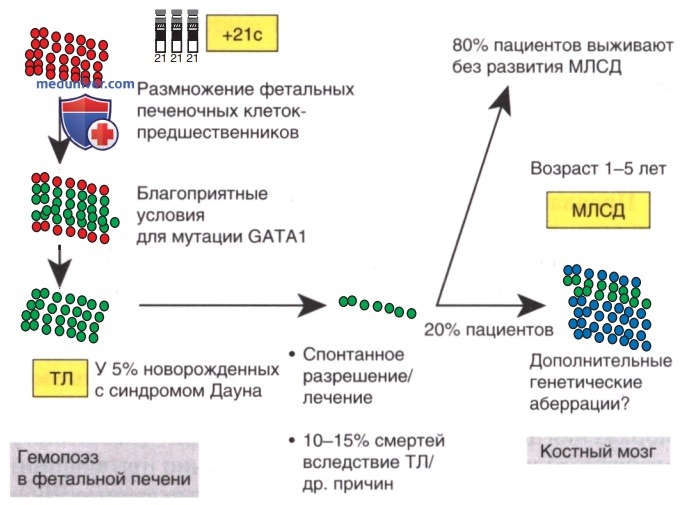
В бластах пациентов с синдромом Дауна, страдающих транзиторным миелопролиферативным заболеванием, а также у больных лейкозом присутствуют мутации гена GATA1 (транскрипционный фактор, контролирующий мегакариопоэз) (рис. 8).
Рисунок 8. Поэтапное развитие миелобластного лейкоза при синдроме Дауна (МЛСД) после транзиторного лейкоза (ТЛ). ТЛ возникает из размножающихся фетальных печеночных клеток-предшественников в результате конституциональной трисомии 21, что обеспечивает благоприятные условия для возникновения приобретенных мутаций гемопоэтического транскрипционного фактора GATA1. В большинстве случаев ТЛ проходит спонтанно, но некоторые дети нуждаются в лечении из-за тяжелых симптомов, обусловленных ТЛ. У 20% детей с ТЛ впоследствии развивается МЛСД, что требует дополнительных действий
г) Хронический миелолейкоз (ХМЛ). ХМЛ — это клональное заболевание кроветворной ткани, на долю которого приходится 2-3% всех случаев детского лейкоза. Примерно 99% случаев характеризуются специфической транслокацией t(9;22)(q34;q11), известной как филадельфийская хромосома, приводящей к формированию гибридного белка BCR-ABL.
Симптомы ХМЛ неспецифичны и могут включать лихорадку, общую усталость, потерю МТ и анорексию. Также может наблюдаться спленомегалия, в результате которой появляется боль в левом верхнем квадранте живота. Подозрение на ХМЛ возникает при высоком уровне лейкоцитов с миелоидными клетками на всех стадиях дифференцировки в периферической крови и костном мозге. Диагноз ХМЛ подтверждается цитогенетическими и молекулярными исследованиями, демонстрирующими наличие характерной филадельфийской хромосомы и перестройки гена BCR-ABL. Эта транслокация, хотя и является характерным признаком ХМЛ, также обнаруживается у небольшого процента пациентов с ОЛЛ.
Заболевание характеризуется начальной хронической фазой, во время которой злокачественный клон продуцирует повышенное количество лейкоцитов с преобладанием зрелых форм, но с повышенным количеством незрелых гранулоцитов. Помимо лейкоцитоза, в ОАК м.б. обнаружены легкая анемия и тромбоцитоз. Как правило, хроническая фаза заканчивается через 3-4 года после начала заболевания, когда ХМЛ переходит в фазу акселерации/фазу «бластного криза». На этом этапе число лейкоцитов резко повышается, а клиническая картина становится неотличимой от клинической картины острого лейкоза. Могут проявляться и др. симптомы, в т.ч. неврологические, обусловленные гиперлейкоцитозом, который вызывает повышение вязкости крови при снижении перфузии ЦНС.
Иматиниб — ЛП, разработанный специально для ингибирования BCR-ABL тирозинкиназы — применялся у взрослых и детей и показал способность вызывать большой цитогенетический ответ у >70% пациентов (см. табл. 1). Опыт работы с детьми показывает его безопасность, а результаты лечения детей этим ЛП сравнимы с результатами у взрослых. Ингибиторы тирозинкиназы второго поколения, такие как дазатиниб и нилотиниб, повысили частоту ремиссий у взрослых и в настоящее время включены в терапию первой линии в этой популяции. Ожидая ответа на ингибитор тирозинкиназы, инвалидизирующие/угрожающие жизни признаки и симптомы ХМЛ можно контролировать во время хронической фазы применением гидроксикарбамида, которая постепенно возвращает количество лейкоцитов к норме. Лечение ингибитором тирозинкиназы является современным стандартом лечения ХМЛ детского возраста.
Хотя в настоящее время этот метод терапии не считается излечивающим, можно наблюдать долгосрочный ответ на терапию, а исследования у взрослых показали, что в отдельных случаях лечение ингибитором тирозинкиназы м.б. прекращено. Роль потенциально излечивающей трансплантации HLA-совместимых стволовых клеток от родственного донора в качестве метода лечения ХМЛ детского возраста является предметом дискуссии.
д) Ювенильный миеломоноцитарный лейкоз (ЮММЛ). ЮММЛ, ранее называемый ювенильным хроническим миелолейкозом, представляет собой клональную пролиферацию гемопоэтических стволовых клеток, которая обычно поражает детей <2 лет. ЮММЛ встречается редко, составляя <1% от всех случаев детского лейкоза. Пациенты с этим заболеванием не имеют филадельфийской хромосомы, характерной для ХМЛ. У пациентов с ЮММЛ наблюдаются сыпь, лимфаденопатия, спленомегалия и геморрагические проявления. ОАК часто показывает повышенное количество лейкоцитов с повышенным содержанием моноцитов, тромбоцитопению и анемию с наличием эритробластов. В костном мозге наблюдается миелодиспластическая картина, причем бласты составляют <20% клеток.
У большинства пациентов с ЮММЛ были обнаружены мутации, приводящие к активации онкогенного пути RAS, включая NRAS, NF1 и PTPN11. Пациенты с нейрофиброматозом 1-го типа и синдромом Нунан (Noonan) имеют предрасположенность к этому типу лейкоза, т.к. у них имеются герминальные мутации, участвующие в передаче сигнального пути RAS. ЮММЛ при синдроме Нунан отличается необычным течением, причем у большинства пациентов происходит спонтанная нормализация состояния. При этом наиболее эффективным методом лечения ЮММЛ для большинства пациентов является трансплантация стволовых клеток, но результаты все еще неудовлетворительные.
е) Младенческий лейкоз. В ~2% случаев лейкоз развивается в возрасте <1 года. В отличие от лейкоза у детей более старшего возраста, у младенцев соотношение ОЛЛ и ОМЛ составляет 2:1. Лейкозные клоны были отмечены в пуповинной крови при рождении до появления симптомов, и в одном случае тот же клон был отмечен в материнских клетках (передача от матери к плоду). Транслокации хромосом могут происходить в/утробно во время фетального гемопоэза, приводя к образованию злокачественных клонов.
Характерными признаками ОЛЛ у детей раннего возраста являются некоторые уникальные биологические особенности и крайне плохой прогноз. В >80% случаев наблюдаются перестройки гена КМТ2А (MLL), обнаруженные в локусе 11q23, большинство из которых являются транслокациями t(4;11). В этой подгруппе пациентов отмечается очень высокая частота рецидивов. У этих пациентов часто наблюдается гиперлейкоцитоз и обширная инфильтрация тканей, приводящая к органомегалии, включая поражение ЦНС.
П/к узелки, известные как гематодерматоз, и тахипноэ, вызванные диффузной легочной инфильтрацией лейкозными клетками, чаще наблюдаются у младенцев, чем у детей более старшего возраста. Морфологически лейкозные клетки обычно представляют собой крупные неправильной формы лимфобласты с фенотипом, «-» для маркера CD10 (общий АГн ОЛЛ) (про-В), в отличие от лейкозных клеток большинства более старших детей с В-клеточным ОЛЛ, «+» для маркера CD10.
У младенцев с перестройками гена КМТ2А (MLL) изучается применение высокоинтенсивных программ XT, включая трансплантацию стволовых клеток, но результаты всех этих программ пока оказались неудовлетворительными. Младенцы с лейкемией при отсутствии перестройки 11q23, имеют тот же прогноз, что и дети с ОЛЛ старшего возраста.
У младенцев с ОМЛ часто наблюдается поражение ЦНС/кожи, они имеют подтип, известный как острый миеломоноцитарный лейкоз. Лечение м.б. таким же, как и у детей с ОМЛ старшего возраста, с аналогичным исходом. Из-за раннего возраста пациентов и агрессивности необходимой им ХТ такие пациенты особенно нуждаются во всесторонней сопроводительной терапии.