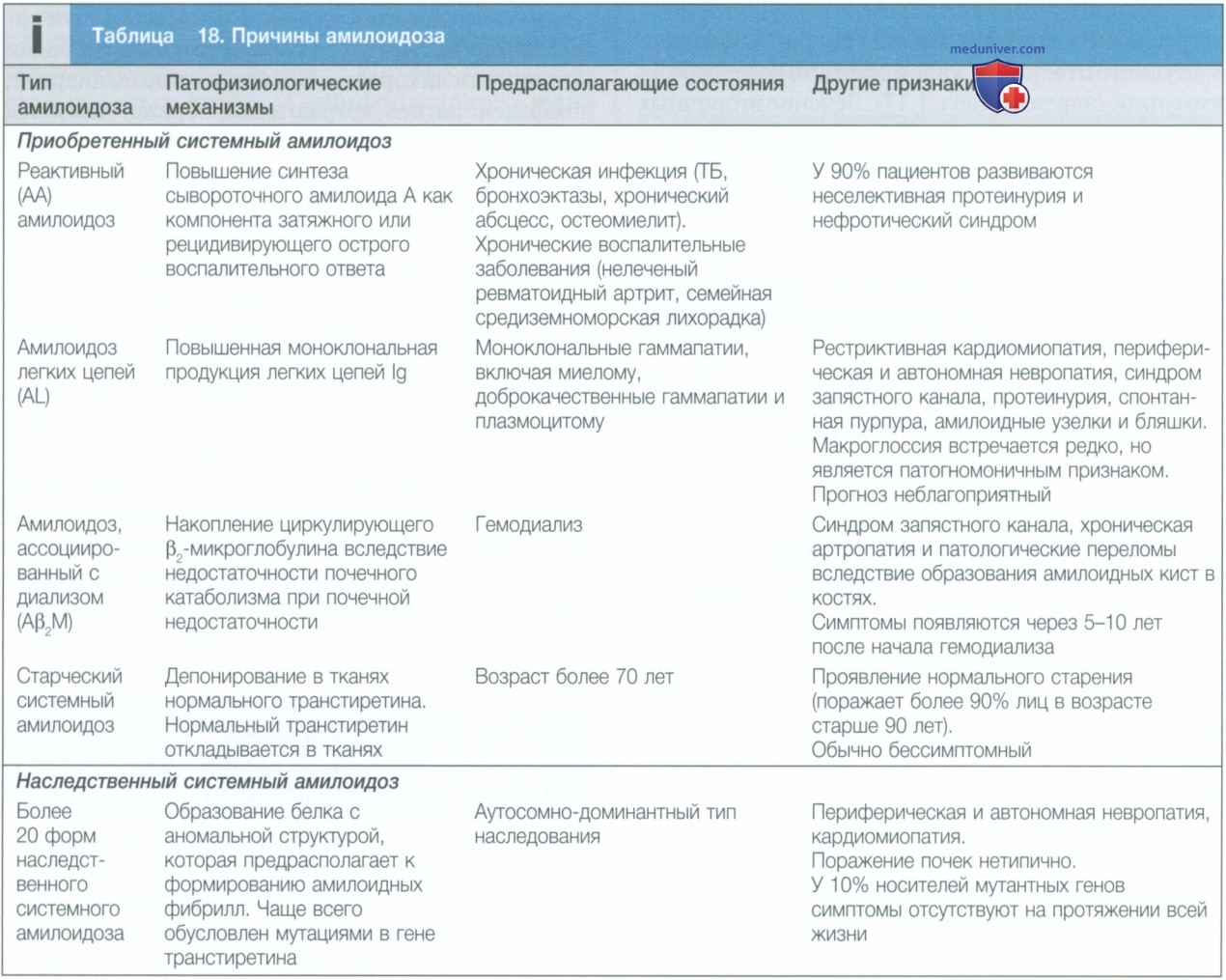
Амилоидоз — группа заболеваний, характеризующихся внеклеточным отложением нерастворимых волокон амилоидных белков в разл. тканях организма.
а) Этиология. Амилоидоз — это заболевание, обусловленное синтезом дефектных белков. Эти неправильно построенные белки инфильтрируют, агрегируют и образуют нерастворимые фибриллы, которые могут влиять на нормальную работу ряда жизненно важных органов.
В номенклатуре амилоидоза различают амилоидоз, развивающийся в результате мутаций белка самих амилоидных фибрилл и амилоидоз, связанный с генетическими мутациями в неамилоидных белках. Эта группа называется наследственными амилоидозами и включает в себя, напр., мутации в генах транстиретина и аполипопротеина А, редко встречающиеся у маленьких детей.
В отличие от наследственных амилоидозов, вторичный амилоидоз А (АА-амилоидоз) развивается у пациентов с хроническими воспалительными заболеваниями. По примерным оценкам, во всем мире ок. 45% всех случаев амилоидоза относятся к АА-амилоидозу. В прошлом причиной большинства случаев АА-амилоидоза были хронические инфекционные заболевания, такие как туберкулез, малярия, проказа и хронический остеомиелит. Теперь, когда эти инфекции эффективно лечатся, более частыми стали др. причины АА-амилоидоза.
С повышенным риском развития АА-амилоидоза ассоциируется ряд хронических воспалительных ревматологических заболеваний, таких как РА, ЮИА и анкилозирующий спондилит, а также наследственные аутовоспалительные заболевания. АА-амилоидоз также ассоциирован с гранулематозными заболеваниями, такими как саркоидоз, муковисцидоз, болезнь Крона, ЗНО (мезотелиома и болезнь Ходжкина), с в/в употреблением наркотиков, бронхоэктазами и ВИЧ. Примерно в 6% случаев АА-амилоидоза не удается установить связь с конкретным заболеванием. AL-амилоидоз (ранее известный как идиопатический амилоидоз или миеломный амилоидоз) чрезвычайно редко наблюдается у детей, встречаясь у людей среднего и старшего возраста.
б) Эпидемиология. У детей в заметном количестве встречается только АА-амилоидоз. Факторы, определяющие вероятность развития амилоидоза как осложнения воспалительного процесса до конца не выяснены, потому что у многих людей с длительно протекающими воспалительными заболеваниями не наблюдается отложения амилоида в тканях, тогда как у некоторых детей с относительно недавним началом заболевания амилоид может образовываться. В развитых странах до начала терапии противоревматическими ЛП, модифицирующими заболевание (DMARDs; англ. diseasemodifying antirheumatic drugs), и биологическими агентами, РА был наиболее распространенным воспалительным заболеванием, ассоциированным с АА-амилоидозом.
Пациенты с длительно протекавшим и плохо контролируемым тяжелым РА с внесуставными проявлениями были группой наибольшего риска развития амилоидоза, и среднее время от появления первых симптомов их ревматологического заболевания до установления диагноза амилоидоза составляло 212 мес. Эффект применения DMARD и биологической терапии при РА-ассоциированном амилоидозе еще только предстоит полностью оценить, но исследования показывают устойчивое снижение количества новых случаев амилоидоза.
ЮИА — еще одно ревматологическое заболевание, ассоциированное с развитием АА-амилоидоза, при этом наиболее часто амилоидоз встречается у пациентов с системным ЮИА, сопровождающимся полиартритом. До начала применения DMARD и биологических ЛП распространенность АА-амилоидоза у пациентов с ЮИА колебалась в пределах 1-10%. Наиболее высокая частота отмечалась у пациентов из Северной Европы, особенно у пациентов из Польши, у которых распространенность амилоидоза составляла 10,6%; наиболее низкая встречаемость описывалась в Северной Америке.
Причины этой разницы в распространенности до конца не изучены, предполагается, что предвзятость отбора пациентов, генетические факторы, а также тенденция к более ранней и более агрессивной терапии в Северной Америке могли сыграть свою роль. АА-амилоидоз выявлялся у пациентов с ЮИА уже через 1 год после постановки диагноза. Так же, как и в случае с РА, выявляемость новых случаев амилоидоза значительно снизилась за последние 20 лет из-за повышения эффективности терапии DMARD и биологическими препаратами.
1. Наследственные аутовоспалительные заболевания — группа заболеваний, характеризующихся приступами, казалось бы, ничем не спровоцированного рецидивирующего воспаления без значительного повышения уровня аутоАТл, АГн-специфических Т-клеток, которые обычно обнаруживаются у пациентов с аутоиммунными заболеваниями. Хотя эти приступы и кажутся ничем не спровоцированными, они часто инициируются стрессом, иммунизацией или травмой. Это позволяет предположить, что важную роль в патогенезе данных процессов играет взаимодействие генетических факторов и факторов окружающей среды.
Несмотря на определенную вариабельность симптомов при аутовоспалительных заболеваниях, к часто наблюдаемым признакам относят лихорадку, кожную сыпь, артрит, серозит и поражение глаз. Воспалительные атаки сопровождаются интенсивными реакциями острой фазы (повышение СОЭ и СРВ) и высокими уровнями сывороточного амилоида А (САА). АА-амилоидоз при этом ассоциирован с некоторыми, но не со всеми наследственными аутовоспалительными заболеваниями.
2. Семейная средиземноморская лихорадка (ССЛ) является наиболее распространенным из менделевских аутовоспалительных заболеваний и наиболее часто встречается в армянской, арабской, турецкой популяциях, а также среди евреев-сефардов. ССЛ — это АуР заболевание, возникающее в результате мутаций гена MEFV, который кодирует синтез белка пирина/маренострина. Мутации MEFV, влияющие на аминокислотные остатки М680 и М694, ассоциированы с ранним началом ССЛ, тяжелым течением заболевания и повышенным риском развития АА-амилоидоза. Пациенты, проживающие в Армении, Турции и арабских странах, имеют повышенный риск развития АА-амилоидоза по сравнению с пациентами с аналогичными мутациями MEFV, живущими в Северной Америке.
Хотя логично было бы предположить, что пациенты с ССЛ, у которых наблюдаются частые тяжелые приступы, будут наиболее подвержены риску развития АА-амилоидоза, это не всегда так. У некоторых пациентов с тяжелыми эпизодами лихорадки в анамнезе амилоидоз так никогда и не развивался, а у др. развивался еще в раннем возрасте. Существует также подгруппа пациентов с ССЛ, называемая фенотипом II. У этих детей АА-амилоидоз развивается еще до первого эпизода лихорадки. В этой группе пациентов наблюдается аналогичная мутация гена MEFV, которая обнаруживается и у больных ССЛ с типичными симптомами.
3. TRAPS связан с мутациями в гене TNFRSF1A, который кодирует рецепторный белок фактора некроза опухоли массой 55 кДа (TNFR1). По приблизительным оценкам, у 14-25% пациентов с TRAPS развивается АА-амилоидоз. Пациенты с мутациями гена TNFRSF1A, который влияет на остатки цистеина, имеют самый высокий риск развития АА-амилоидоза. Считается, что эти остатки цистеина участвуют в формировании дисульфидных связей, важных для сборки TNFR1, и что разрушение этих связей влияет на синтез белка.
Мутации в гене NLRP3 (также известном как CIAS1, ген криопирин-ассоциированного периодического синдрома 1) вызывают 3 клинически отличающихся друг от друга заболевания: семейный холодовой аутовоспалительный синдром, синдром Макл-Уэллса и мультисистемное воспалительное заболевание новорожденного, также известное как хронический детский неврологический кожно-суставной синдром. Мутации гена NLRP3 наследуются по аутосомно-доминантному типу или как мутации de novo у пациентов с наиболее тяжелым течением заболевания. У меньшей части пациентов обнаруживаются соматические мутации в NLRP3.
Семейный холодовой аутовоспалительный синдром чаще всего является наименее тяжелой формой среди криопиринопатий и редко ассоциируется с развитием АА-амилоидоза. Синдром Макл-Уэллса проявляется лихорадкой, миалгиями, артралгиями, уртикарной сыпью и прогрессирующей нейросенсорной тугоухостью. АА-амилоидоз довольно часто встречается при синдрое Макл-Уэллса, поражая до трети пациентов. Мультисистемное воспалительное заболевание новорожденного/хронический детский неврологический кожно-суставной синдром — самая тяжелая форма криопиринопатии. Исторически ок. 20% пациентов с этим заболеванием умирали, не достигнув совершеннолетия, но при нынешних методах лечения многие живут дольше.
У некоторых пациентов с мультисистемным воспалительным заболеванием новорожденногос возрастом развивается АА-амилоидоз, хотя и не так часто, как у пациентов с синдромом Макл-Уэллса, что, возможно, связано с меньшей продолжительностью жизни этих пациентов.
4. Гипер-IgD синдром — еще одно аутовоспалительное заболевание, которое дебютирует в раннем детском возрасте, проявляясь ознобом, высокой ТТ, болями в животе, лимфаденопатией и иногда сыпью. Гипер-IgD синдром — это АуР-заболевание, вызванное мутациями, ведущими к потере функции, в гене MVK, который кодирует фермент мевалонаткиназу. Тяжелые мутации гена MVK, которые полностью устраняют активность фермента, выявляются у пациентов с мевалоновой ацидурией, у которых наблюдается рецидивирующая лихорадка, дисморфические особенности и задержка в развитии. Гипер-IgD синдром — ассоциированные мутации представляют собой мутации с более мягким снижением функции. Маркеры воспаления, включая сывороточный амилоид А (САА), повышаются во время приступов и могут оставаться повышенными в межприступный период.
АА-амилоидоз при гипер-IgD синдром встречается редко, но сообщения о таких случаях были.
Хорошо известно о риске развития АА-амилоидоза у пациентов с болезнью Крона, хотя он и встречается реже, чем при наследственных синдромах периодической лихорадки. АА-амилоидоз встречается у 1% пациентов в США и у 3% пациентов из Северной Европы. В то же время АА-амилоидоз у больных язвенным колитом встречается крайне редко, с примерной распространенностью 0,07%. Такие пациенты, как правило, имеют длительный анамнез агрессивного, плохо контролируемого заболевания, хотя есть сообщения об амилоидозе у пациентов с хорошо контролируемыми маркерами воспаления.
Наследственный амилоидоз, связанный с транстиретином, — это АуД-заболевание с переменной пенетрантностью и началом во 2-3-м десятилетии жизни. Болезнь вызывается более чем 120 одиночными или двойными мутациями в гене TTR. Проявления ее включают в себя нейропатию (семейную амилоидотическую полинейропатию: моторную, сенсорную, вегетативную), семейную амилоидную кардиомиопатию, нефропатию и поражения глаз.
в) Патогенез. Отложение амилоидных фибрилл типа АА является результатом длительного воспалительного процесса, который приводит к синтезу дефектного АА-амилоидного белка и его отложению в тканях. Белком-предшественником фибрилл при АА-амилоидозе является аполипопротеин, называемый сывороточным амилоидом А. САА экспрессируется 3 разными генами, локализованными на хромосоме р15.1. САА1 и САА2 — две изоформы этого аполипопротеина, которые являются реактантами острой фазы, синтезируемыми печенью и способными образовывать амилоид. САА вырабатывается в ответ на провоспалительные цитокины, такие как IL-1, IL-6 и ФНО-α, и его уровень может возрастать более чем в 1000 раз во время воспаления. Было высказано предположение, что САА играет роль хемоаттрактанта и в метаболизме липидов.
Эта теория подтверждается первоначальным отложением амилоида в органах, которые являются основными участками метаболизма липидов и холестерина, таких как почки, печень и селезенка. Примерно 80% секретируемых САА1 и САА2 связаны с липопротеинами.
В норме САА, секретируемый печенью, полностью разрушается макрофагами. САА имеет длину в 104 аминокислоты и в основном секретируется в структуре а-спирали. По не совсем понятным причинам пациенты с АА-амилоидозом имеют дефект, приводящий к неполной утилизации САА и накоплению его промежуточных продуктов. У таких пациентов САА переносится в лизосому, где с-концевая часть белка САА расщепляется, позволяя оставшемуся белку формироваться в конфигурацию |3-складчатого листа. Депонированный амилоид содержит только 66-76 аминокислот по сравнению с 104 в секретируемом САА. Эти отщепленные фрагменты полимеризуются и образуют фибриллы, которые откладываются во внеклеточном пространстве и связывают протеогликаны и другие белки, такие как сывороточный амилоид Р. Эти фибриллы затем становятся устойчивыми к протеолизу и откладываются в тканях внутренних органов.
Образование АА-амилоида может быть ассоциировано с рядом факторов риска. Ген, кодирующий САА1, имеет полиморфизм, наличие которого приводит к повышению риска развития АА-амилоидоза в 3-7 раз. Пациенты европеоидной расы с РА, ЮИА или аутовоспалительными заболеваниями с генотипом SAAa/a имеют повышенный риск развития амилоидоза. В этой группе пациентов аллель SSA1γ связан со сниженным риском амилоидоза. Интересно отметить, что у японских пациентов картина обратная: генотип SAAa/a связан со сниженной вероятностью развития амилоидоза, а генотип SAA1γ ассоциирован с повышенным риском.
г) Клинические проявления. Хотя поражение органов может быть различным, АА-амилоидоз чаще всего поражает почки, 90% пациентов имеют определенную степень поражения почек. У некоторых пациентов первым симптомом может быть необъяснимая протеинурия. Если основное воспалительное заболевание не контролируется или его диагностика проводится недостаточно эффективно, может развиться нефротический синдром и почечная недостаточность. Медиана выживаемости после установления диагноза составляет 133 мес. Пациенты с более высоким уровнем САА имели значительно более высокий риск смерти, чем пациенты с более низким уровнем.
Поражение ЖКТ наблюдается у 20% пациентов и обычно проявляется хронической диареей, ЖКК, болями в животе и мальабсорбцией. При биопсии часто обнаруживается поражение семенников (87%). Относительно редкие находки, связанные с АА-амилоидозом, включают в себя анемию, амилоидный зоб, гепатомегалию, спленомегалию, поражение надпочечников и легких. Такие ткани как сердце, язык и кожа поражаются редко.
д) Диагностика. Диагноз амилоидоза устанавливается путем биопсии, которая обнаруживает наличие белков амилоидных фибрилл в пораженных тканях. Исследуемые ткани включают в себя почки, прямую кишку, жировую прослойку БП и десны. Амилоидные отложения состоят из гомогенного эозинофильного материала, который окрашивается красителем конго красный и демонстрирует при этом патогномоничное «двойное лучепреломление яблочно-зеленого цвета» в поляризованном свете. Гист. и генетическое исследование полезно при транстиретиновом амилоидозе.
е) Лабораторные показатели. У пациентов с АА-амилоидозом обычно наблюдается повышение уровней реактантов острой фазы и высокий уровень Ig. В США специальные лабораторные исследования на АА-амилоид недоступны по коммерческим причинам, но в др. странах уровень САА можно контролировать и использовать для оценки ответа на лечение.
ж) Лечение. Не существует однозначно утвержденной терапии АА-амилоидоза, и, следовательно, основным подходом является агрессивное лечение основного воспалительного или инфекционного заболевания, что позволит снизить уровень САА. По мере разработки новых методов лечения основного заболевания появляются и новые данные, свидетельствующие о снижении заболеваемости АА-амилоидозом. Колхицин эффективен не только для борьбы с приступами ССЛ, но и для предотвращения развития амилоидоза, ассоциированного с ССЛ. Дети с ССЛ, гомозиготные по мутации M694V в MEFV, подвержены большему риску развития амилоидоза и должны находиться под пристальным наблюдением.
В отличие от АА-амилоидоза, ассоциированного с ССЛ, АА-амилоидоз, ассоциированный с др. аутовоспалительными заболеваниями (включая TRAPS, криопи-рин-ассоциированный периодический синдром и редко гипер-IgD синдром) и хроническими ревматологическими заболеваниями (ЮИА, РА и анкилозирующий спондилит), не реагирует на колхицин. Хотя АА-амилоидоз, ассоциированный с ЮИА, может реагировать на хлорамбуцил, прием этого ЛП связан с повреждением хромосом и риском последующего развития ЗНО. Распространение биологических ЛП, направленных против провоспалительных цитокинов в терапии РА, ЮИА, спондилоартропатий и наследственных аутовоспалительных заболеваний, по-видимому, влияет и на факторы риска развития АА-амилоидоза.
Класс ЛП, называемых ЛП ингибиторы ФНО-а (иФНО), играет главенствующую роль в терапии РА и др. аутоиммунных заболеваний. Как при аутоиммунных, так и при аутовоспалительных заболеваниях, сопряженных с развитием АА-амилоидоза, имеются сообщения, подтверждающие эффективность иФНО в замедлении прогрессирования амилоидоза. Побочные эффекты иФНО включают реактивацию туберкулеза и гепатита В, поэтому перед их назначением следует провести тщательный скрининг. Кроме того, у пациентов, принимающих иФНО, было отмечено образование разл. АТл, аутоАТл и развитие аутоиммунных заболеваний. С особой осторожностью следует назначать и ФНО пациентам с СН или демиелинизирующим заболеванием в анамнезе, поскольку их применение может вызвать обострение основной сердечной и неврологической патологии. Путь IL-1 является мишенью для многих биологических ЛП, используемых при аутоиммунных и аутовоспалительных заболеваниях.
Существует 3 доступных антагониста IL-1 — это анакинра (антагонист рецептора IL-1), рилрнацепт (растворимый рецептор-ловушка IL-1) и канакинумаб (полностью гуманизированное моноклональное АТл IgG1 к IL-1β длительного действия). Разл. ингибиторы IL-1 успешно замедляют прогрессирование АА-амилоидоза, а в некоторых случаях терапия даже приводит к регрессу амилоид-ассоциированной протеинурии.
Было показано, что тоцилизумаб, АТл против рецептора IL-6, в эксперименте ослабляет АА-амилоид и обращает вспять развитие АА-амилоидоза, осложняющего ССЛ, ЮИА и РА. Недавнее исследование с использованием динатрия эпродисата у пациентов с АА-амилоидозом не смогло достичь своей основной конечной цели, заключавшейся в замедлении прогрессирования почечной недостаточности до терминальной стадии.
Транстиретиновый амилоидоз лечат с помощью трансплантации печени и транстиретин-стабилизирующих ЛП.
з) Прогноз. Терминальная стадия почечной недостаточности является основной причиной смерти у 40-60% пациентов с амилоидозом, при этом среднее оставшееся время жизни с момента постановки диагноза составляет 2-10 лет. Согласно результатам крупномасштабного исследования 374 пациентов с АА-амилоидозом, факторы, ассоциированные с неблагоприятным прогнозом, включают пожилой возраст, сниженный уровень альбумина сыворотки, терминальную стадию почечной недостаточности уже при постановке диагноза и длительное повышение уровня САА в сыворотке. Повышенное значение САА было главным фактором риска развития терминальной стадии почечной недостаточности и смерти от АА-амилоидоза.
и) Профилактика. Основным средством профилактики АА-амилоидоза является лечение основного воспалительного или инфекционного заболевания, что приводит к снижению уровня САА, и уменьшает риск отложения амилоида. Несмотря на то что длительность латентного периода от начала воспаления (основного заболевания) до появления первых клинических признаков АА-амилоидоза может варьировать и зачастую латентный период является продолжительным, отложение и накопление амилоида может протекать очень быстро.