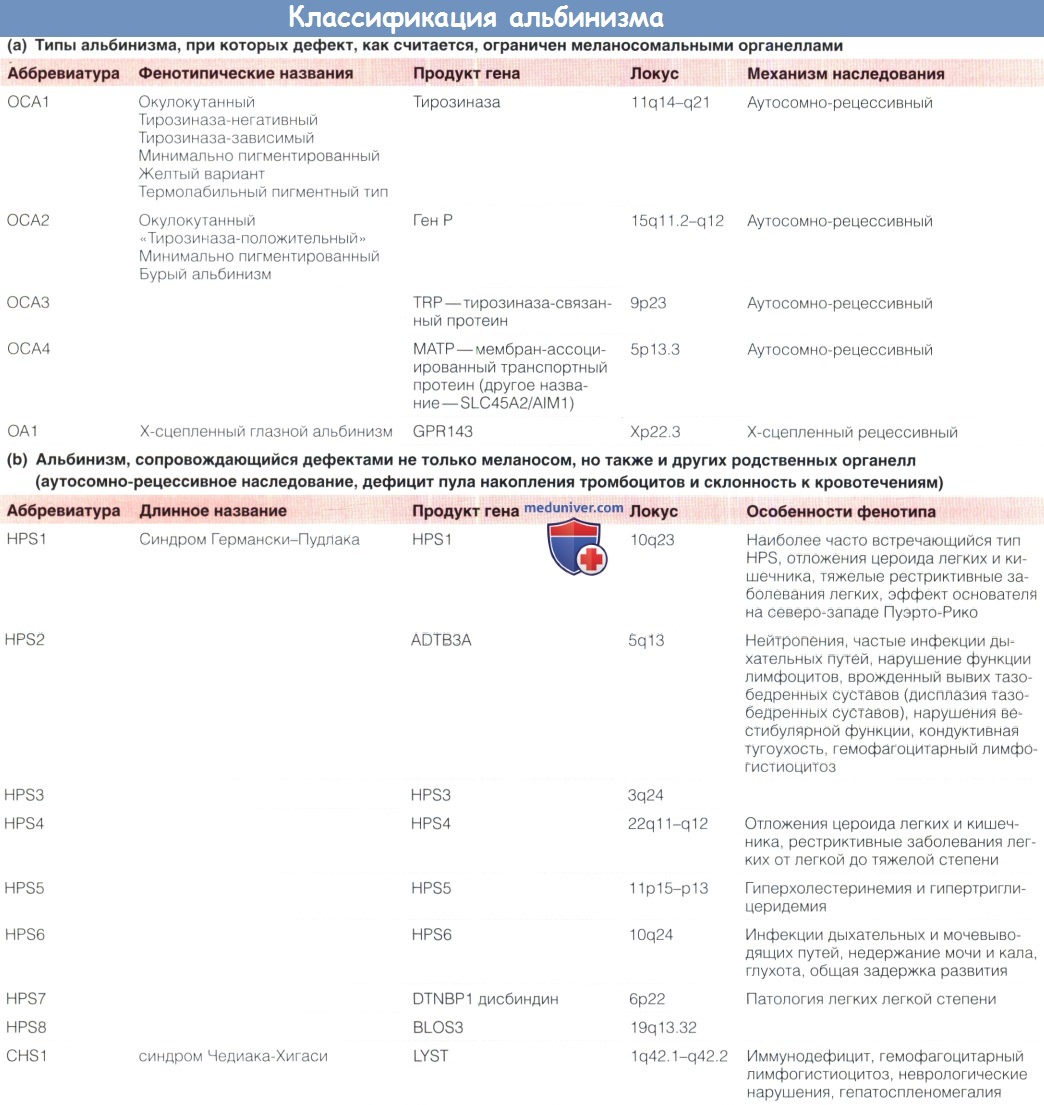
Тирозиназа — ключевой фермент метаболизма меланина, ее недостаточность вызывает альбинизм у многих животных. Хотя это также частая причина альбинизма у человека (окулокутанный альбинизм 1 типа, oculocutaneous albinism type 1 — ОСА 1), причиной альбинизма также могут быть дефекты других звеньев метаболизма.
Протеин ОСА2Р стабилизирует pH меланосомы, что создает условия для образования меланина. Тирозиназа взаимодействует с тирозиназа-связанным протеином (tyrosinase related protein) TYRP1 (его дефект вызывает окулокутанный альбинизм 3 типа — ОСА3); мутация, приводящая к дефекту одного, нарушает созревание и стабильность другого. Мутация только TYRP1, вероятно, не приводит к развитию полного фенотипа с нистагмом и аномалиями хиазмы. Протеин МАТР (ОСА4) важен для нормального функционирования тирозиназы и созревания меланосом.
В выработке меланина и его транспорте из органеллы у млекопитающих принимают участие многие другие белки, некоторые из которых не специфичны для меланосом, а содержатся также в родственных лизосомам органеллах (LROs).
Удобнее всего подразделять альбинизм на состояния, при которых дефект продукта гена вызывает изменения только в пределах меланосом, и заболевания, при которых также возникают аномалии родственных органелл. В первом случае у пациента наблюдаются непрогрессирующие аномалии глаз и недостаточность фотопротекции, тогда как в последнем случае могут развиваться тяжелые системные нарушения.
Альбинизм встречается во всех этнических группах, но его распространенность неодинакова из-за «эффекта основателя». Встречаемость HPS1 в Пуэрто-Рико составляет 1:1800, в других популяциях она может снижаться даже до одного случая на миллион жителей. Примерно один из семидесяти жителей Северной Европы является носителем одной аномальной аллели ОСА1. ОСА2 преобладает среди народностей Южной и Центральной Африки, где часто встречается делеция 2,7, приводящая к утрате одного экзона гена.
ОСА4 редко встречается за пределами Японии, где он составляет до 25% альбинизма. Генотипы ОСА1, ОСА2, ОСА3, ОСА4,ОА1 и HPS1 вызывают более 90% случаев альбинизма в большинстве популяций и в совокупности определяют встречаемость альбинизма в США около 1 на 17000.
а) Наследование альбинизма и взаимодействие продуктов генов. Все формы альбинизма наследуются по аутосомно-рецессивному механизму, за исключением ОА1, наследуемого по Х-сцепленному механизму. На экспрессию дефектного менделирующего гена влияет взаимодействие с другими генами, регулирующими обмен меланина.
В будущем, возможно, удастся разработать ген-специфическое лечение. В настоящее время дифференциация генотипов, не сопровождающихся поражением других органелл, выполняется с целью генетического консультирования. Взаимодействие генов приводит к появлению огромного количества вариантов пигментации у здоровых людей и у альбиносов. При альбинизме фенотип варьирует в зависимости от этнической группы.
Если больной с альбинизмом вследствие наличия у него двух мутантных аллелей ОСА1 будет иметь детей от альбиноса, имеющего две мутантных аллели ОСА2, все их дети будут носителями генов обоих типов заболевания, но альбинизм не разовьется ни у одного из них, поскольку эти два типа альбинизма являются различными неаллельными состояниями. Если у индивида имеется нуль-мутация одной аллели ОСА1 и вторая аллель, наблюдается полиморфизм со снижением активности фермента, изменения глаз не развиваются, за исключением случаев наличия также мутации MITF, гена, участвующего в том же регуляторном пути.
Суммарное действие двух генов одного регуляторного пути является примером дигенного наследования. В Пуэрто-Рико из-за «эффекта основателя» отмечается высокая частота не одного, а сразу двух различных типов HPS; встречаются гомозиготы по одному типу — носители другого типа; у таких пациентов развивается более тяжелый фенотип. Индивиды с ОСА2, у которых образуется некоторое количество меланина, могут иметь каштановые или рыжие волосы. Их пигментация определяется генотипом — состоянием гена рецептора меланокортина (MC1R).
б) Смежные с альбинизмом генные синдромы. Делеция участка X-хромосомы в зоне Хр22.3 вызывает развитие ОА1 и нарушение функции других генов этой зоны. Фенотип больных мужчин варьирует в зависимости от локализации и объема делеции.
Гены и локусы этой зоны и соответствующая симптоматика:
• NLGN4X Нейроглин 4: аутизм, нарушения обучаемости
• STS Стероидная сульфатаза: ихтиоз
• ARSE Арилсульфатаза Е: точечная хондродисплазия
• ОА1 альбинизм
• KALI синдром Кальмана (гипогонадотропинемический гипогонадизм и аносмия)
• LECD эпителиальная дистрофия роговицы Лиша
• OASD альбинизм и поздно развивающаяся глухота
• SHOX гомеобокс низкого роста
в) Редкие типы альбинизма. Окулоцеребральный синдром с гипопигментацией (синдром Кросса, OMIM 257800): симптоматика включает в себя серебряно-серые волосы, гипопигментацию кожи, нистагм, помутнение роговицы, микрофтальмию, перипапиллярные пигментированные «рубцы», задержку психомоторного развития, спастичность и экстрапирамидные движения. Описан случай немой электроретинограммы.
Окулоцеребральный гипопигментационный синдром Preus (OMIM257790): проявления включают в себя задержку роста, долихоцефалию, катаракты, нистагм, прозрачную радужку, высокое готическое небо, мелкие широко посаженные зубы, генерализованную гипопигментацию, задержку психомоторного развития и гипохромную анемию.
Нейроэктодермальный меланолизосомальный синдром Элехальде (OMIM256710): серебристые волосы и высокая частота летальных неврологических эпизодов. Возможно, аллелен синдрому Griscelli.
Взаимодействие гена Р с другими генами. И при синдроме Ангельмана и при синдроме Прадера-Вилли гипопигментация кожи, волос и радужки развивается вследствие делеции гена Р. Альбинизм развивается при мутации второго гена Р. Синдром Прадера-Вилли характеризуется гипотонией в младенческом возрасте, гиперфагией и ожирением, гипогонадизмом, умственной отсталостью, маленьким ростом, маленькими кистями и стопами.
Причиной заболевания является дефект длинного плеча 15 хромосомы в зоне гена Р, возникающий вследствие делеции участка отцовской хромосомы, мутации центра контроля геномного импринтинга, хромосомной транслокации или однородительской дисомии материнской хромосомы 15. У многих больных отмечается просвечивающая радужка и гипопигментация кожи.
Синдром Ангельмана характеризуется тяжелой задержкой развития, нарушениями речи, атаксией, микроцефалией, судорожными припадками и частыми приступами немотивированного смеха. По результатам исследования ЗВП описаны аномалии путей хиазмы, не сопровождающиеся другими глазными симптомами альбинизма. Синдром Ангельмана развивается вследствие делеции участка материнской хромосомы в той же зоне, дефект которой вызывает синдром Прадера-Вилли, однородительской дисомии (в отличие от синдрома Прадера-Вилли — отцовской хромосомы), нарушения хромосомного импринтинга или мутации гена UBE3A.
г) Путаница в терминологии альбинизма. Следует отказаться от термина «частичный альбинизм», поскольку он используется как в отношении заболеваний, например пьебалдизма, при которых отмечается отсутствие меланоцитов на отдельных участках кожи и не развиваются характерные для альбинизма изменения зрительного пути, так и для обозначения тех случаев истинного альбинизма, когда некоторое количество пигмента все же присутствует.
Термином «глазной альбинизм» обозначают как альбинизм с Х-сцепленным наследованием, ОА1, при котором аномальные меланосомы присутствуют не только в пределах глаза, так и аутосомно-рецессивные формы альбинизма, при которых гипопигментация не выражена.
Выделение именных синдромов на основании старых описаний фенотипа может вводить в заблуждение, поскольку в настоящее время их этиопатогенез изучен более глубоко: например, HPS2 сопровождается нейтропенией и, как и CHS1, нарушением функции лимфоцитов; CHS1 связан с недостаточностью тромбоцитов, а не только с нарушениями иммунитета.