Синонимы синдрома Риза. S. Krause — Reese. S. Krause. S. Reese—Blodi. Дисплазия Reese. Дисплазия сетчатой оболочки. Peтинальная дисплазия. Врожденная энцефало-офтальмическая дисплазия (Krause).
Определение синдрома Риза. Комбинированное врожденное преимущественно семейное заболевание с дисплазией сетчатой оболочки и двусторонними пороками развития головного мозга и других органов.
Авторы. Reese Algernon В. — современный американский офтальмолог, Нью-Йорк. Blodi Frederick С. — современный американский офтальмолог, Нью-Йорк. Krause Arlington С. — современный американский офтальмолог. По-видимому, первый случай описал в 1891 г. Bernheimer; авторами последующих сообщений были Dotsch (1899), А. С. Krause (1946) и Francois (1947). Reese и Blodi (1950) впервые привели комплексное описание синдрома и отграничили данный тип дисплазии сетчатой оболочки от других типов.
Симптоматология синдрома Риза:
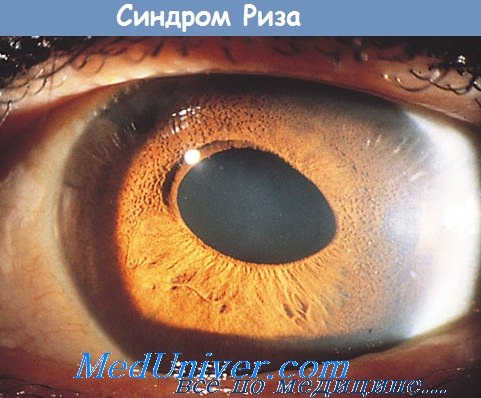
1. Глаза: двусторонний порок развития сетчатой оболочки (различной степени выраженности) в форме недостаточной дифференцировки ее слоев, ретинальной дис- и гиперплазии, что гистологически проявляется развитием складок и «розеток». В связи с этим развиваются вторичная отслойка сетчатки, псевдоглиома, двусторонняя микрофтальмия всех степеней, уплощение передней камеры; сохраняются первичные отделы стекловидного тела и частично — зрачковая мембрана.
Радужная оболочка искривлена вследствие наличия задних синехий. Реже встречается помутнение хрусталика. Резко выраженное снижение остроты зрения вплоть до амавроза. Нередко встречаются также колобома и кистозные позадиглазные образования (орбитальные кисты), а также аномалии зрительного нерва (сохранение оболочки стекловидного тела).
2. Центральная нервная система: гидроцефалия, энцефалоцеле, менингоцеле, общая гипоплазия головного мозга, дисплазия мозжечка (по типу S. Arnold—Chiari) и ствола мозга с соответствующими множественными выпадениями и психическими дефектами.
3. Легкие: аплазия или гипоплазия (уменьшение числа) легочных долей, первичные ателектазы, диафрагмальная грыжа.
4. Пищеварительная система: пилоростеноз, атрезия двенадцатиперстной кишки, аплазия или гипоплазия желчного пузыря, гепатомегалия, подвижная слепая кишка, омфалоцеле.
5. Костная система: расщепление нижней челюсти и неба, микрогнатия, пороки развития стоп, синили полидактилия, пороки развития позвонков со сколиозом и т. д.
6. Сердце: дефект перегородки, незаращение овального отверстия, аортальный порок.
7. Мочеполовая система: кистозные почки, кистозные яички, крипторхизм, фимоз.
Этиология и патогенез синдрома Риза неизвестны. Генетическое (комплексная мутация гена) или эпигенетическое расстройство с многочисленными пороками развития (в глазе сетчатая оболочка не образует вторичного corpus vitreum; она не расположена на наружном слое вторичного глазного пузыря и сохраняется с первичным стекловидным телом, которое не подвергается инволюции). В одном случае с резко выраженными изменениями была выявлена трисомия 13 (Saraux с соавторами, 1964).
Дифференциальный диагноз. S. Terry (см.). S. Gregg (см.). Глиома сетчатой оболочки. Гидрофтальмия. Врожденная катаракта. Ретинобластома. Дисрафия.